








Simultaneous multiple target detection in real-time loop-mediated isothermal amplification
Abstract
Loop-mediated isothermal amplification (LAMP) is a rapid and reliable sequence-specific isothermal nucleic acid amplification technique. To date, all reported real-time detection methods for LAMP have been restricted to single targets, limiting the utility of this technique. Here, we adapted standard LAMP primers to contain a quencher-fluorophore duplex region that upon strand separation results in a gain of fluorescent signal. This approach permitted the real-time detection of 1–4 target sequences in a single LAMP reaction tube utilizing a standard real-time fluorimeter. The methodology was highly reproducible and sensitive, detecting below 100 copies of human genomic DNA. It was also robust, with a 7-order of magnitude dynamic range of detectable targets. Furthermore, using a new strand-displacing DNA polymerase or its warm-start version, Bst 2.0 or Bst 2.0 WarmStart DNA polymerases, resulted in 50% faster amplification signals than wild-type Bst DNA polymerase, large fragment in this new multiplex LAMP procedure. The coupling of this new multiplex technique with next generation isothermal DNA polymerases should increase the utility of the LAMP method for molecular diagnostics.
Sequence-specific isothermal nucleic acid amplification techniques represent a growing sector of molecular diagnostics, offering rapid, sensitive detection without the need for thermal cycling equipment as required for the PCR. Furthermore, isothermal techniques typically provide comparable or better detection limits compared with PCR but in a fraction of the reaction time (1,2). These methods have particular interest to field or point-of-care molecular diagnostics due to advantages in efficiency, cost, and instrumentation (3). In contrast to PCR, which denatures double-stranded DNA (dsDNA) with heat, isothermal amplification techniques typically use enzymatic activity to provide strand separation of dsDNA. Sequence specificity is provided by oligonucleotide primers that anneal to the target sequence and are extended by a strand displacing DNA polymerase. Several techniques require multiple enzymes to work in concert [e.g., strand displacement amplification (SDA), helicase dependent amplification (HDA), and isothermal and chimeric primer-initiated amplification of nucleic acids (ICAN)], but loop-mediated isothermal amplification (LAMP) provides sequence-specific amplification using only a strand-displacing DNA polymerase (techniques reviewed in References 1 and 2). In addition to the DNA polymerase, LAMP uses four core primers (FIP, BIP, F3, and B3) recognizing six distinct sequence regions on the target (Figure 1), with two primers containing complementary sequence in order to create loop structures that facilitate exponential amplification (4). The use of multiple target sequence regions confers a high degree of specificity to the reaction. Two additional primers, termed loop primers, can be added to increase reaction speed, resulting in six total primers used per target sequence (5). The LAMP reaction rapidly (completed in as little as 5 min) generates amplification products as multimers of the target region in various sizes and is substantial in total DNA synthesis (>10 µg, >50× PCR yield) (4–6).
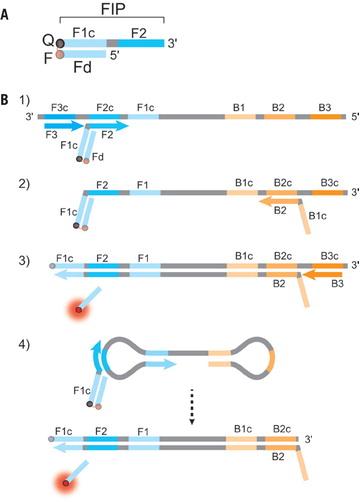
(A) Schematic depiction of a DARQ probe, with a 5′-quencher FIP (F1c + F2 sequence) annealed to a 3′-fluorophore Fd. The quencher and fluorophore moieties are represented by Q and F, respectively. (B) Outline of DARQ LAMP reactions, with core LAMP primers FIP (F1c + F2), BIP (B1c + B2), F3 and B3, and the DARQ oligonucleotide, Fd (Q, black; F, red). For clarity, LoopF and LoopB primers are not shown. (1) LAMP initiates at the F2c sequence of the target, with the Fd probe quenched through annealing to Q-FIP. This new strand is displaced by upstream synthesis from the F3 primer. (2) The BIP primer anneals to the B2c site in the newly synthesized strand. (3) Synthesis from the primer annealed to the B2c sequence displaces the Fd probe. This releases the quenching resulting in a gain of signal. The newly synthesized strand is displaced by extension from the B3 primer. (4) The resulting structure undergoes exponential amplification in the LAMP reaction. Subsequent initiations at FIP give rise to additional release of Fd, resulting in exponential signal detection.
Measurement of the LAMP product is typically performed by fluorescence detection of dsDNA with an intercalating or magnesium-sensitive fluorophore (4,7), bioluminescence through pyrophosphate conversion (8), turbidity detection of precipitated magnesium pyrophosphate (9,10), or even visual examination through precipitated Mg2P2O7 or fluorescence (11,12). These methods are robust and familiar, and visual methods are ideal for use in field diagnostics, but detect total DNA amplification in a reaction and are thus limited to detection of a single target. As isothermal techniques are further adopted as diagnostic tools, the ability to detect multiple targets in a single sample will be important. Currently, quantitative PCR (qPCR) enables probe-specific multiplex detection and the ability to perform tests with an internal standard for definitive negative results. However, qPCR probes require extensive design and optimization for use and may not effectively translate to the LAMP reaction (13–16). Previous studies have demonstrated methods of multiplex LAMP detection, although they have been limited in capability. A variety of methods have used end point analysis, through agarose gel (17,18) or pyrosequencing (19) methods, but these do not allow real-time detection and require further processing and instrumentation. In addition, the sensitivity of LAMP reactions to carryover contamination is so great that manufacturer recommendations (Eiken Chemical, Tokyo, Japan) suggest not opening LAMP reaction vessels, or doing so in separate facilities with separate equipment, furtherdecreasing the desirability of post-LAMP manipulation. Previous real-time methods use nonspecific quenching, either through loss-of-signal guanine quenching (20) or gain-of-signal fluorescence using labeled primers and an intercalating dye (21). These methods can be less sensitive, and nonspecific quenching limits the selection of fluorophores available for multiplexing. Our approach was to develop a gain-of-signal, target-specific LAMP detection that is easily implemented and not limited in design or sensitivity.
The LAMP forward and back interior primers (FIP and BIP) contain 5′ flaps (Figure 1; F1c sequence) that, upon synthesis and displacement, will anneal to a complementary downstream region (F1). We chose this region for development of detection probes, as it is inherent to LAMP and contains sequence that is specific to each target, precluding any need for probe sequence optimization. LAMP also requires a strand displacing DNA polymerase (typically Bst DNA polymerase, large fragment), a component we utilize for detection through strand displacement. Using previously designed LAMP primers as a basis, we synthesized the FIP modified at the 5′ end with either a dark quencher or fluorophore. For probe creation, we annealed oligonucleotides complementary to the flap region (F1c) with a 3′ dark quencher or fluorophore spectrally overlapping with the fluorophore or dark quencher of the FIP (Figure 1A). This duplex primer maintains its function as a LAMP primer, but upon synthesis from the reverse direction the flap duplex is separated, resulting in detection of amplification by release of quenching (Figure 1B). A similar method using a short nested probe rather than amplification primer was applied to qPCR (22). Our method, which we termed detection of amplification by release of quenching or DARQ, is adaptable for any LAMP reaction and requires no additional probe design or testing, merely synthesis of a 5′-modified FIP and a 3′-modified complementary probe (termed Fd). Similar quencher:fluorophore duplex primers have been previously used in isothermal amplification, including LAMP with suggested use for mulitplexing, but to date no demonstration of real-time, multiplex LAMP has been achieved in this fashion (23–25). Here, we demonstrate the use of this method for LAMP detection in single and multiplex reactions, detecting up to four distinct LAMP targets in a single reaction.
Materials and methods
LAMP primers were designed either manually or using PrimerExplorer V4 (Eiken Chemical). Sequences can be found in Supplementary Table S1, and all synthetic oligonucleotide primers, Q-FIP, and Fd were synthesized by Integrated DNA Technologies (Coralville, IA, USA). The dark quencher was either Iowa Black FQ or RQ, and fluorophores used were 6-FAM, HEX, ROX, Cy5, and Cy5.5, each corresponding to one of five channels in a CFX96 Real Time System (Bio-Rad Laboratories, Hercules, CA, USA), used for performing LAMP reactions (Supplementary Table S2).
Q-FIP:Fd duplexes were annealed by heating 50 µM Q-FIP and 50 µM Fd to 98°C and slowly cooling the mixture to room temperature. LAMP reactions with Bst 2.0 DNA polymerase or Bst 2.0 WarmStart DNA polymerase (New England Biolabs, Ipswich, MA, USA) were performed in 1× Isothermal Amplification Buffer (New England Biolabs): 20 mM Tris-HCl (pH 8.8, 25°C), 10 mM (NH4)2SO4, 50 mM KCl, 2 mM MgSO4, 0.1% Tween-20 supplemented to 8 mM MgSO4, and 1.4 mM each of dATP, dCTP, dGTP, and dTTP. LAMP reactions withstandard Bst DNA polymerase, LF (New England Biolabs) used a similar buffer based on ThermoPol DF (New England Biolabs), identical in composition as above but with 10 mM KCl in place of 50 mM KCl. LAMP reactions contained: 1.6 µM FIP (or 0.8 µM FIP and 0.8 µM Q-FIP:Fd), 1.6 µM BIP, 0.2 µM F3 and B3, 0.4 µM LoopF 10 mM KCl in place of 50 mM KCl. LAMP reactions contained: 1.6 µM FIP (or 0.8 µM FIP and 0.8 µM Q-FIP:Fd), 1.6 µM BIP, 0.2 µM F3 and B3, 0.4 µM LoopF and LoopB, and 0.64 U/µL Bst DNA polymerase, LF, Bst 2.0 DNA polymerase, or Bst 2.0 WarmStart DNA polymerase. For multiplex reactions, total primer concentrations were kept to those described for the standard LAMP reaction, but with each set representing 1/n of the total, where n is the number of targets. Bacteriophage λ genomic DNA (5 ng/reaction) and HeLa genomic DNA (100 ng/reaction) were from New England Biolabs, Escherichia coli genomic DNA (5 ng/reaction) was from Affymetrix (Santa Clara, CA, USA), and Caenorhabditis elegans genomic DNA (82.5 ng/reaction) was purified using standard procedures. Reactions were performed at 65°C in triplicate, and all presented Ct values represent an average ± standard deviation.
Results and discussion
In DARQ, the standard FIP primer is modified with a 5′ quencher and annealed to an F1c-complementary detection probe, Fd (Figure 1). To test this method for LAMP detection, we designed four sets of LAMP primers with Q-FIP and accompanying Fd probes, each with a different fluorophore and quencher pair. These targeted genes from different organisms and genome complexities: E. coli dnaE (Iowa Black RQ/Cy5); C. elegans lec-10 (RQ/ROX); human cystic fibrosis transmembrane conductance regulator (CFTR; FQ/6-FAM); and human BRCA1 (RQ/Cy5.5). Additionally, we adapted a set of LAMP primers for bacteriophage λ DNA (5) with the quencher and fluorophore positions reversed (5′-HEX FIP/3′-FQ) to examine any effect of quencher/fluorophore location. Q-FIP:Fd duplexes were made for each primer set, and LAMP reactions performed using duplex FIP primers.
Figure 2 shows LAMP amplification of dnaE from E. coli genomic DNA with DARQ detection using Bst DNA polymerase, large fragment or standard and WarmStart versions of Bst 2.0 DNA polymerase. Release of quenching can be seen as an increase in Cy5 signal, and use of all three polymerases resulted in robust amplification and signal (Figure 2A). The reaction was found to proceed faster when using either the standard or WarmStart versions of Bst 2.0 DNA polymerase as compared with wild-type Bst DNA polymerase (Figure 2 and Supplementary Figure S1). Therefore, Bst 2.0 DNA polymerase was used in all subsequent experimentation. Use of 5′ modified FIP primer alone had no effect on LAMP, but we observed some relative inhibition of amplification when Q-FIP:Fd duplexprimer completely replaced the standard FIP primer (Supplementary Figure S1). This inhibition was significantly reduced through the use of equimolar standard FIP primer and Q-FIP:Fd duplex (Supplementary Figure S2). This effect was likely due to faster target generation with FIP and easier incorporation of duplex FIP during exponential amplification, and using equimolar amounts maintains rapid threshold detection with high fluorescence signal amplitude, important for detecting amplification in multiplex reactions.

LAMP primers were specific for the dnaE gene of E. coli (5 ng genomic DNA per reaction) as described in the Materials and methods section. The FIP primer was labeled with Iowa Black RQ and the DARQ probe had a 3′ Cy5. (A) Three Bst DNA polymerases provided robust signal, with faster amplification seen for Bst 2.0 and Bst 2.0 WarmStart DNA polymerases. (B) Duplicate reactions to (A) were performed after 2 h preincubation at room temperature, with substantial negative effects on amplification time and yield for non-WarmStart polymerases, but identical performance was observed with WarmStart Bst 2.0. The reactions were performed as described in the Materials and methods section.
A convenient feature of LAMP F1c regions is that they are typically 20–25 bases long and are selected by Primer Explorer to feature Tm greater than 60°C. Thus by the nature of the LAMP primer design, the F1c:Fd duplex remains stably annealed in our reactions at 63°–65°C, and no signal is observed in the absence of strand-displacing DNA polymerase. However, if nonstandard primer sequences are required, DARQ reactions can simply be performed at lower temperatures to accommodate less stable duplexes. The F1c regions described here range in Tm from 61°–74°C (Supplementary Table S1), and all perform DARQ LAMP reactions at 60°–65°C, indicating that use of F1c:Fd duplexes does not limit primer design considerations. As described above, we also tested a primer pair with fluorophore and quencher positions switched on FIP and Fd. Use of this reverse orientation primer set (λ) resulted in no difference in amplification detection efficiency (Figure 3A and Figure 4) or duplex primer inhibition levels (data not shown), thus we can conclude that DARQ primers can be synthesized with either a 5′ quencher or fluorophore FIP if necessary to accommodate limited modified oligonucleotide synthesis chemistry.

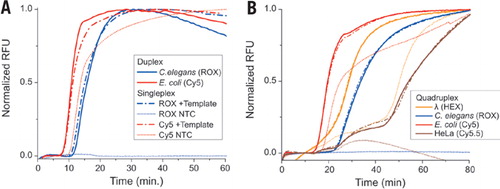
DARQ detection of two distinct LAMP amplifications in the same reaction, either with similar (A) or disparate (B) amplification times. Data are graphed as normalized fluorescence units to account for differences in fluorophore quantum yield. (C) Maintenance of amplification for an internal standard (C. elegans lec-10/ROX; 82.5 ng C. elegans genomic DNA) with varying amounts of test target DNA (10 pg–100 ng HeLa; ∼3–30,000 copies). The reactions were performed as described in the Materials and methods section.

Detection of three (A) and four (B) LAMP targets in the same reaction using distinct sets of DARQ primers. A slight increase in detection time accompanies the decrease in specific primer concentration due to added target sequences. Data are graphed as normalized fluorescence units to account for differences in fluorophore quantum yield. The reactions were performed as described in the Materials and methods section.
A common problem in nucleic acid amplification reactions is undesired activity from DNA polymerases during room temperature reaction setup (26,27). This activity may not be a problem when performing a limited number of samples set up by hand, but it can result in irreproducibility for medium- or high-throughput or automated workflows. To test whether LAMP and DARQ detection may be affected by preincubation at room temperature, identical LAMP reactions were set up and performed with or without a 2-h preincubation at 25°C. Both Bst DNA polymerase large fragment and Bst 2.0 DNA polymerase were negatively impacted by the 2-h preincubation, likely due to low levels of DNA polymerase activity at 25°C (Figure 2). In PCRs, this problem is typically overcome through the use of “hot start” DNA polymerases. Bst 2.0 WarmStart DNA polymerase provides this functionality for isothermal amplification techniques through an engineered aptamer that inhibits polymerase activity below 50°C (28), and its efficacy in DARQ is shown in Figure 2. Bst 2.0 WarmStart maintains identical amplification profiles whether LAMP is performed immediately after set up (Figure 2A) or after 2 h preincubation at 25°C (Figure 2B). This benefit accommodates room temperature reaction setup, but otherwise does not affect Bst 2.0 performance, so all subsequent data are from standard Bst 2.0.
With DARQ detection validated, we next sought to extend the method to multiple targets detected in a single reaction. Figure 3 presents fluorescence curves from LAMP reactions containing two distinct, complete primer sets and their corresponding genomic DNA targets. For multiple target amplification, we maintained the total amount of primer in a standard LAMP reaction; using each set at full concentration resulted in suboptimal performance as concentration increased (i.e., total primer concentration was kept to 4.4 µM regardless of the number of templates, with each primer set adjusted by 1/n, where n is number of targets in the reaction). As shown in Figure 3, DARQ robustly detects distinct targets in a single LAMP reaction. Curves shown are normalized to maximum fluorescence signal in that channel to account for differences in the quantum yield of various fluorophores. The detection provided robustsignal for each target regardless of the speed of their independent amplification, which will vary based on the nature of the primers, templates, and target copy number. Despite the quicker amplification reaching exponential phase sooner, the slower amplification was observed to proceed unaffected, obviating the need for consideration of amplification speed when multiplex targets are selected (Figure 3B). This independent nature of DARQ reactions allows maintenance of sensitivity when performed with multiple targets, as shown in Figure 3C, where FAM-CFTR is detected to ∼2.9 copies of HeLa genomic DNA (10 pg) in the same reaction as robust LAMP for ROX-lec-10 (82.5 ng C. elegans DNA, ∼7.6 × 105 copies). Thus a robust LAMP standard curve can be generated across a copy number range of Target 1 (here, CFTR) while Target 2 (lec-10) is detected simultaneously. Amplification of the constant target remains unchanged (all 5 ROX Ct values 11.8 ± 0.03 min) across the copy number range of the variable target providing a reliable positive control (Figure 3C). This property allows LAMP to be performed with an internal standard, an important consideration for diagnostic applications. Figure 3C demonstrates DARQ performance at low copy numbers, but high copy numbers are also reliably detected, as seen in Figure 2 (5 ng E. coli genomic DNA, ∼106 copies) and Figure 3A (5 ng λ DNA, ∼108 copies). Use of DARQ detection thus imposes no limitation to the sensitivity of the LAMP reaction. Similarly, the dynamic range of LAMP is unaffected by duplex DARQ, which maintains robust detection from 10–108 copies.
DARQ can easily be extended to three and four target reactions (Figure 4), again with total primer concentration constant and each set adjusted for number of targets. Reducing primer concentration 3- or 4-fold does accordingly increase time to reach threshold. This drop in time was consistent, making template quantification reliable, and the reaction times were still rapid with Bst 2.0 DNA polymerase. The multiplexed reactions display robust amplification of three (Figure 4A) or four (Figure 4B) targets, with loss of signal amplitude accompanying decreased concentration of the fluorophore-containing primer. This fluorescence decrease emphasizes the need for bright fluorophores, with high quantum yield and appropriate spectral matching with the fluorescence detection channels. In DARQ reactions, ROX gave the best results (>20,000 background-subtracted fluorescence counts in single-plex reactions), likely due to a 15 nm wider detection channel. Cy5, Cy5.5, and HEX gave similarly high signal (10,000–15,000), but 6-FAM (<5,000) gave lower fluorescence signal that became difficult to distinguish from background fluorescence when diluted 3- or 4-fold for multiplex reactions. Thus DARQ is accommodating of any fluorophore that can be quenched and detected, but for detecting more than three targets simultaneously brighter dyes are preferred.
An essential consideration for amplification techniques or any diagnostic methodology is proper identification of positive reactions from false positive or negative reactions, either from nontemplate (amplification of primers without template DNA) or nonspecific amplification (amplification of a nontarget DNA present in the sample), although the latter is unlikely in LAMP due to the use of six sequence-specific primers per target. LAMP and other techniques can result in nontemplate amplification due to several factors, including the nature of LAMP primers and the reaction conditions, which include high concentrations of primer (4.4 µM total) and magnesium (8 mM). The degree of this nontemplate amplification is determined by the specific primer sequences (27), and we demonstrate this in DARQ in Figure 5, which shows a high degree of nontemplate amplification for one primer set (E. coli dnaE), an intermediate level for another (bacteriophage λ), and none for two sets (C. elegans lec-10 and human BRCA1). Importantly for diagnostic testing and multiplexing, the reactions containing many different templates maintained their level of specificity, i.e., the positive and nontemplate (nonspecific) signal was identical for a primer set whether no template or several nonspecific genomic DNAs were present. Figure 5B shows this specificity, with each primer set amplifying target identically in the presence of either a single genomic DNA (dashed lines) or four genomic DNAs (solid lines). Each negative control reaction (dotted lines) was carried out with three nonspecific genomic DNAs, and no effect was seen on nontemplate amplification, e.g., C. elegans lec-10 primers gave no signal without any genomic DNA (Figure 5A) or in the presence of bacteriophage λ, human, and E. coli DNA (Figure 5B). This allows accurate detection and quantification of a target (e.g., human BRCA1) in the presence of high levels of other genomic DNAs (e.g., 100 ng bacteriophage λ, 82.5 ng C. elegans, and 100 ng E. coli). Although we did not test clinical samples with DARQ, LAMP has been widely used for specimen diagnostics, afforded by an increased tolerance to typical contaminants and inhibitors (29). There is no obvious reason that DARQ detection would not effectively translate into diagnostics using clinical samples, however this needs to be experimentally verified.
Panel A shows a primer set with a high nontemplate signal, E. coli dnaE primers, and a primer set with no nontemplate signal, C. elegans lec-10 primers. In the absence of DNA template, the E. coli primer set gave an amplification signal (red dotted line), whereas the C. elegans primer set gave no signal (blue dotted line). In the presence of appropriate DNA template, each primer set gave a robust positive signal that was unchanged whether performed as singleplex (dashed lines) or duplex (solid lines) reaction. Panel B shows real-time DARQ reactions for bacteriophage λ, C. elegans, E. coli, and HeLa (BRCA1) DNA. The primers were used in singleplex (dashed lines) or quadruplex (solid lines) reactions. The nontemplate control reactions (dotted lines) contained the three unrecognized genomic DNAs appropriate for each primer set. Nontemplate signals were high for E. coli and bacteriophage λ primer sets but negative for C. elegans and HeLa primer sets. Note that the positive signals for each primer set were the same in the singleplex and quadruplex reactions, indicating that there was no spurious amplification due to the presence of three nonspecific genomic DNAs in each quadruplex reaction.
DARQ detection provides a method for multiplex detection of LAMP amplification with no need for additional primer optimization or probe design, requiring only the use of a 5′ modified LAMP primer (FIP) with complementary detection oligonucleotide (Fd). This detection methodology may be extendable to other nucleic acid amplification techniques by the addition of a duplex “tail” to a primer in the reaction. Upon extension from the opposite direction, the duplex would be displaced with detection upon release of quenching. Our detection method is a simple extension of LAMP to accommodate robust, target-specific, and multiplex detection. As molecular diagnostics become more prominent and accepted in healthcare, the ability to detect multiple targets and use of internal controls will further the utility and flexibility of LAMP.
Acknowledgments
The authors thank Dr. Gregory Lohman for helpful discussion, Drs. Andrew Gardner, Gregory Lohman, and Jennifer Ong for critical reading of the manuscript, and New England Biolabs, Inc. for financial and material support.
Competing interests
The authors are employed by New England Biolabs, Inc., manufacturer of the described enzymes.
Supplementary data
To view the supplementary data that accompany this paper please visit the journal website at: www.future-science.com/doi/suppl/10.2144/0000113902
References
- 1. . 2008. Nucleic acid isothermal amplification technologies: a review. Nucleosides Nucleotides Nucleic Acids 27:224–243.
- 2. . 2011. Isothermal DNA amplification in bioanalysis: strategies and applications. Bioanalysis 3:227–239.
- 3. . 2011. Point-of-care nucleic acid testing for infectious diseases. Trends Biotechnol. 29:240–250.
- 4. . 2000. Loop-mediated isothermal amplification of DNA. Nucleic Acids Res. 28:E63.
- 5. . 2002. Accelerated reaction by loop-mediated isothermal amplification using loop primers. Mol. Cell. Probes 16:223–229.
- 6. . 2001. Loop-mediated isothermal amplification reaction using a nondenatured template. Clin. Chem 47:1742–1743.
- 7. . 2009. Colorimetric detection of loop-mediated isothermal amplification reaction by using hydroxy naphthol blue. BioTechniques 46:167–172.
- 8. . 2010. Novel bioluminescent quantitative detection of nucleic acid amplification in real-time. PLoS One 5:e14155.
- 9. . 2004. Real-time turbidimetry of LAMP reaction for quantifying template DNA. J. Biochem. Biophys. Methods 59:145–157.
- 10. . 2001. Detection of loop-mediated isothermal amplification reaction by turbidity derived from magnesium pyrophosphate formation. Biochem. Biophys. Res. Commun 289:150–154.
- 11. . 2008. Loop-mediated isothermal amplification (LAMP) of gene sequences and simple visual detection of products. Nat. Protocols 3:877–882.
- 12. . 2011. Adaptation of a visualized loop-mediated isothermal amplification technique for field detection of Plasmodium vivax infection. Parasit. Vectors 4:115.
- 13. . 1991. Detection of specific polymerase chain reaction product by utilizing the 5′—3′ exonuclease activity of Thermus aquaticus DNA polymerase. Proc. Natl. Acad. Sci. USA 88:7276–7280.
- 14. . 2008. Twenty-five years of quantitative PCR for gene expression analysis. BioTechniques 44:619–626.
- 15. 2001. DNA probes using fluorescence resonance energy transfer (FRET): designs and applications. BioTechniques 31:1106–1121.
- 16. 2006. A-Z of Quantitative PCR. International University Line, La Jolla, CA.
- 17. . 2010. A single fluorescence-based LAMP reaction for identifying multiple parasites in mosquitoes. Exp. Parasitol 125:179–183.
- 18. . 2010. Development of a multiplex loop-mediated isothermal amplification (mLAMP) method for the simultaneous detection of white spot syndrome virus and infectious hypodermal and hematopoietic necrosis virus in penaeid shrimp. Aquaculture 311:94–99.
- 19. . 2012. Multiplex LAMP detection by sequence-based barcodes coupled with NEase-mediated Pyrosequencing. Anal. Chem 84:3758–3763.
- 20. . 2010. Methylation-specific loop-mediated isothermal amplification for detecting hypermethylated DNA in simplex and multiplex formats. Clin. Chem 56:1287–1296.
- 21. . 2010. Homogenous, real-time duplex loop-mediated isothermal amplification using a single fluorophore-labeled primer and an intercalator dye: Its application to the simultaneous detection of Shiga toxin genes 1 and 2 in Shiga toxigenic Escherichia coli isolates. Mol. Cell. Probes 24:190–195.
- 22. . 1995. Quantitative PCR using the AmpliSensor assay, p. 193–202. In C.W. Dieffenbach and G.S. Dveksler (Eds.), PCR Primer—A Laboratory Manual. CSH Laboratory Press, Cold Spring Harbor, NY.
- 23. . 2009. Sequence-specific detection method for reverse transcription, loop-mediated isothermal amplification of HIV-1. J. Med. Virol 81:966–972.
- 24. . 2006. Molecular Zipper: a fluorescent probe for real-time isothermal DNA amplification. Nucleic Acids Res. 34:e81.
- 25. . 2011. FRET-based assimilating probe for sequence-specific real-time monitoring of loop-mediated isothermal amplification (LAMP). Biological Engineering Transactions 4:81–100.
- 26. . 1994. TaqStart antibody: “hot start” PCR facilitated by a neutralizing monoclonal antibody directed against Taq DNA polymerase. BioTechniques 16:1134–1137.
- 27. . 2011. Optimization of turn-back primers in isothermal amplification. Nucleic Acids Res. 39:e59.
- 28. 1997. The joys of in vitro selection: chemically dressing oligonucleotides to satiate protein targets. Curr. Opin. Chem. Biol 1:10–16.
- 29. . 2007. Tolerance of loop-mediated isothermal amplification to a culture medium and biological substances. J. Biochem. Biophys. Methods 70:499–501.



