








Simultaneous amplification of 5′ and 3′ cDNA ends based on template-switching effect and inverse PCR
Abstract
A new approach for simultaneously amplifying the 5′ and 3′ ends of a desired cDNA is described. The method combines the template-switching effect with inverse PCR, which generates the flanking 5′ and 3′ regions of a certain cDNA with very low or no background. It requires only minimal amounts of total RNA for the synthesis of first-strand cDNA, while the same cDNA can be used to amplify flanking sequences of any cDNA species present in the sample. This method is reliable and easy to perform, which is very useful for isolating cDNA species of rare transcripts.
Introduction
Rapid amplification of cDNA ends (RACE) is an efficient approach for obtaining full-length cDNA when only partial sequences are available (1–3). The principle of RACE is that an anchor sequence (to be used as the PCR primer binding template) is added to the end of the cDNA, followed by a series of hemi-nested anchored PCRs to generate the flanking sequences. Normally 5′ and 3′ RACE are performed separately. Because only one gene-specific primer is used, traditional 5′ and 3′ RACE often generate a high level of nonspecific amplification especially for the 5′ RACE. Several strategies, such as the CapSelect, step-out PCR, and solid phase cDNA synthesis, have been developed to eliminate background problems (2–6). These strategies depend on the so-called CapFinder approach (first described by Clontech on its web site), which is based on the ability of the Moloney murine leukemia virus (MMLV) reverse transcriptase to add 2–4 cytosine residues to the 3′ end of newly synthesized cDNAs upon reaching the 5′ end (cap region) of the messenger RNA (mRNA). When an oligonucleotide with 3–4 G residues (named the T-S oligo) at its 3′ end is present in the reverse transcriptase reaction, the T-S oligo can base pair with the 2–4 C residues of the newly synthesized cDNA. Reverse transcriptase then switches templates (from the mRNA to the oligonucleotide) and continues transcribing the oligonucleotide, thus attaching the complementary oligonucleotide sequence to the 3′ terminus of the cDNA (Figure 1). The advantage of the template-switching procedure is its simplicity, in that it avoids using a set of enzymatic reactions after the completion of first strand cDNA synthesis involved in most RACE approaches, such as homopolymeric tailing and ligation anchored tailing. However, during the reverse transcriptase reaction, the T-S oligo is free to anneal not only to the oligonucleotide stretch at the 3′ end of the cDNA, but also potentially to anywhere on the RNA acting as a primer for reverse transcription, which could cause heavy background if the T-S PCR primer is used in a subsequent PCR. PCR suppression (4) and solid phase cDNA synthesis protocols (5) could not totally eliminate the problems arising from contamination of the primers used for cDNA synthesis or/and nonspecific cDNAs generated by the CapFinder primer, because similar to other RACE methods, these approaches used only one gene-specific primer. Thus, PCR with two gene-specific primers should be a better way to solve the problem. Inverse PCR has been used in the amplification of flanking unknown sequences by using two primers pointing away from the known sequences (7–9).
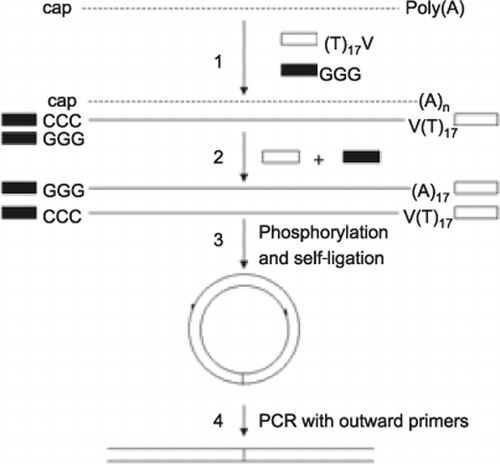
First-strand cDNA synthesis is initiated at the poly(A)-tail of messenger RNA (mRNA) using the oligo(dT)-anchor primer (step 1). The CapFinder primer (Table 1, T-S primer containing G residues at the 3′ most part) could base pair with the newly added 2–4 C residues of the synthesized cDNA, which in turn functions as a template for the reverse transcriptase. As a result, the newly synthesized cDNA contains the sequence complementary to the T-S primer sequence at the 3′ end and the oligo(dT)-anchor primer sequence at the 5′ end. The cDNA is amplified in PCR using the T-S PCR primer and PCR anchor primer (Table 1, step 2). The PCR products are phosphorylated and self-ligated (step 3), and used as templates for inverse PCR (step 4). Inverse PCR is performed with one pair of outward primers specific to the known sequence region of a double-stranded cDNA. The inverse PCR product represents the 5′ and 3′ joining regions of an interest gene.
Here we report a modified reverse transcription inverse PCR approach for performing 5′ and 3′ RACE simultaneously. This approach combines the advantages of the template-switching effect (simplicity in obtaining full-length cDNAs and attaching an oligonucleotide sequence to the 3′ terminus of the full-length cDNAs) and inverse PCR (generation of a specific target product).
Materials and methods
The organisms, genes, and oligonu-cleotides used in this study are listed in Table 1. Total RNA was isolated using TRI Reagent® (Molecular Research Center, Cincinnati, OH, USA). All oligonucleotides were synthesized from Invitrogen (Carlsbad, CA, USA). First strand cDNA was synthesized with 1 µg total RNA. The reaction conditions were as follows: 10 pmol anchor oligo(dT) was annealed to 1 µg total RNA in a volume of 10 µL RNase-free water, by heating the mixture at 70°C for 10 min, followed by cooling on ice for 5 min. Transcription was then initiated by mixing the annealed RNA with 200 U PowerScript™ Reverse Transcriptase (Clontech, Mountain View, CA, USA) in a final volume of 20 µL containing 1× first-strand buffer, 10 mM dithiothreitol (DTT), 10 pmol T-S primer, and 1 mM each of dATP, dGTP, dCTP, and dTTP. The reaction was incubated at 42°C for 90 min. Aliquots (1–2 µL) of this reaction were subjected to a Platnum® Pfx DNA polymerase PCR system (Invitrogen) in 50 µL with the PCR anchor primer and T-S PCR primer (Table 1). The double-stranded cDNA products were purified with GlassMax® spin cartridges (Invitrogen). For phosporylation, 0.1 µg purified double-stranded cDNA was diluted to a concentration of 2 ng/µL in 1× ligation buffer, and 5 U T4 polynucleotide kinase (Fermentas, Hanover, MD, USA) were added. The reaction was incubated for 30 min at 37°C. The kinase was inactivated at 70°C for 10 min followed by a 2-fold dilution of the reaction mixture with 1× T4 ligase buffer to replenish the ATP. The intramolecular circularization was initiated by the addition of T4 DNA ligase (Fermentas) to a concentration of 4 U/100 µL, and the reaction was incubated at 4°C for 16 h.
 |
The AccuPrime™ Taq DNA Polymerase System (Invitrogen) was used to amplify target 5′ and 3′ cDNA ends. Outward primers were designed based on the sequence information of each gene. The PCR amplification was conducted in a 50-µL volume containing 1 ng self-ligated cDNAs, 1× PCR buffer, 100 µM each of dATP, dGTP, dCTP, and dTTP, and 0.2 µM primers. Nested PCR was performed with 1 µL PCR product from the first round of amplification and a pair of nested primers. Both rounds of amplification used the same cycle conditions: 5 min at 94°C, followed by 32 cycles of 94°C for 25 s, 60°C for 30 s, and 68°C for 1 min/ kb. For comparison, 5′ and 3′ RACE were performed using a 5′/3′ RACE kit (Roche Diagnostics GmbH, Mannheim, Germany).
Results and discussion
The strategy of this approach is illustrated in Figure 1. To assess the efficiency of the approach, we tried to simultaneously amplify the 5′ and 3′ regions of carotenoid hydroxylase cDNA (chy, GenBank® accession no. AF162276) from a green microalga Haematococcus pluvialis. Two outward specific primers (Table 1, primers 1 and 2) were designed to amplify a 1-kb 5′ and 3′ joining region of the chy gene with self-ligating double-stranded cDNAs as templates (Figure 1). The PCR amplification failed to generate distinct fragments (Figure 2A, lane 2). However, a hemi-nested PCR (with primers 1 and 3) amplified two distinct fragments (Figure 2A, lane 3). Sequencing data of the fragments revealed that they belonged to the joining sequences of the 5′ and 3′ regions of the chy with different length of the 3′ regions (data not shown), indicating the existence of different chy species.

Figure 2. Application of inverse PCR to amplification of the 5′ and 3′ joining regions of chy (A) and 14-3-3 (B) cDNA from Haematococcus pluvialis. First-strand cDNA was synthesized starting from total RNA with oligo(dT) anchor and T-S primers. Two pairs of outward primers specific to each of the genes were used: (A and B) lanes 2, first round of PCR; lanes 3, second round of PCR (nested PCR); lanes 1, 100-bp DNA Ladder Plus (Fermentas).
In order to determine if the same self-ligating cDNAs could be used to amplify other cDNA species, we attempted to clone an undocumented 14–3-3 cDNA from the alga with the same self-ligating cDNAs. One pair of outward primers (Table 1, primers 4 and 5) was designed based on a partial 14–3-3 cDNA sequence (unpublished data). The PCR amplification failed to generate distinct fragments (Figure 2B, lane 2). However a nested PCR (with primers 5 and 6) generated a robust band (Figure 2B, lane 3), which was identified as the 5′ and 3′ regions of a putative 14–3-3 cDNA (GenBank accession no. AY973204). These results indicated that our approach is efficient in obtaining both 5′ and 3′ cDNA ends simultaneously, and the same self-ligating double-stranded cDNAs could be used to amplify different cDNA species present in the sample.
To compare the efficiency of our method in isolating desired genes with that of traditional 5′ and 3′ RACE, we used an undocumented rare transcript of the pcs (phytochelatin synthase) gene from Sesbania rostrata (a plant species). As seen in Figure 3, the first round PCR of 5′ and 3′ RACE could not generate specific fragments (Figure 3 A, lanes 2 and 4). The second round PCR (hemi-nested PCR) of 5′ RACE of pcs also failed to generate the desired product (Figure 3A, lane 3). Similar to traditional 5′ and 3′ RACE, our approach failed to generate the desired product in the first round PCR (Figure 3B, lane 2). However, the second round PCR generated robust DNA fragments (Figure 3B, lane 3). Sequence data revealed that the fragment contains the complete 5′ region including the start codon and the 3′ region of a putative pcs cDNA (GenBank accession no. DQ013293). These results showed that our method is also efficient in obtaining full-length cDNA sequences of rare transcripts from which traditional RACE failed to generate clonable products.

(A) Lanes 2 and 3, first and second rounds of 5′ RACE; lanes 4 and 5, first and second rounds of 3′ RACE. (B) Lanes 2 and 3, first and second rounds of inverse PCR. (A and B) Lanes 1, 100-bp DNA Ladder Plus.
In this study, we have developed an alternative strategy in simultaneous amplification of 5′ and 3′ cDNA ends. The method has several advantages over the former reverse transcription inverse PCR approach (9). First, our method is much simpler and requires only a minimal amount of total RNA (about 1 µg). Second, a nested PCR in our approach greatly increases its sensitivity and specificity, making inverse PCR more likely to be successful. As a result, the 5′ and 3′ regions of a desired cDNA could be reliably amplified simultaneously. The approach described here is very useful for isolating unknown cDNA when only minimal sequence of a gene is available, when 5′ and 3′ regions of a certain cDNA require isolation, and when other RACE approaches fail to capture rare transcripts.
Acknowledgments
We would like to thank MissYu Binyuang and Dr. Xu Zengfu from Zhongshan University for providing the total RNA sample of Sesbania rostrata and the partial sequence information of the pcs gene. This work was supported by a grant from the Research Grants Council of Hong Kong and the Outstanding young Researcher Award of the University of Hong Kong.
Competing Interests Statement
The authors declare no competing interests.
References
- 1. . 1988. Rapid production of full-length cDNAs from rare transcripts: amplification using a single gene-specific oligonulceotide primer. Proc. Natl. Acad. Sci. USA 85:8998–9002.
- 2. . 1999. Inverse single-strand RACE: an adapter-independent method of 5′ RACE. BioTechniques 27:656–658.
- 3. . 2003. 3′ RACE walking along a large cDNA employing tiered suppression PCR. BioTechniques 34:750–756.
- 4. . 1999. Amplification of cDNA ends based on template-switching effect and step-out PCR. Nucleic Acids Res. 27:1558–1560.
- 5. . 2000. A simple and reliable 5′-RACE approach. Nucleic Acids Res. 28:22–e96.
- 6. . 2000. 5′ RACE by tailing a general template- switching oligonucleotide. BioTechniques 29:1192–1195.
- 7. . 1988. Genetic application of an inverse polymerase chain reaction. Genetics 120:621–625.
- 8. . 1988. A procedure for in vitro amplification of DNA segments that lie outside the bound aries of known sequences. Nucleic Acids Res. 16:8186–8188.
- 9. . 1990. A simple method for direct cloning cDNA sequence that flanks a region of known sequence from total RNA by applying the inverse polymerase chain- reaction. Nucleic Acids Res. 18:1922–1992.



