








Chromatin immunoprecipitation assay
Abstract
Association between proteins and DNA is crucial for many vital cellular functions such as gene transcription, DNA replication and recombination, repair, segregation, chromosomal stability, cell cycle progression, and epigenetic silencing. It is important to know the genomic targets of DNA-binding proteins and the mechanisms by which they control and guide gene regulation pathways and cellular proliferation. Chromatin immunoprecipitation (ChIP) is an important technique in the study of protein-gene interactions. Using ChIP, DNA-protein interactions are studied within the context of the cell. The basic steps in this technique are fixation, sonication, immunoprecipitation, and analysis of the immunoprecipitated DNA. Although ChIP is a very versatile tool, the procedure requires the optimization of reaction conditions. Several modifications to the original ChIP technique have been published to improve the success and to enhance the utility of this procedure. This review addresses the critical parameters and the variants of ChIP as well as the different analytical tools that can be combined with ChIP to enable better understanding of DNA-protein interactions in vivo.
Introduction
DNA-protein interactions play a key role in the regulation of important cellular functions including gene transcription, DNA replication and recombination, repair, segregation, chromosomal stability, cell cycle progression, and epigenetic silencing. The 3-dimensional structure of chromatin is maintained by the binding of histones and other regulatory proteins to the DNA. It is vital to know how DNA-binding proteins affect the functioning of any particular gene and to identify which particular protein binds to a specific DNA sequence in vivo. Earlier methods devised to study the DNA-protein interactions involve in vitro methods, which are not within the context of the cell, and thereby have limited utility. Chromatin immunoprecipitation (ChIP) has become a very widely used technique for determining the in vivo location of binding sites of various transcription factors (1–3), histones (4,5), and other proteins (6). Because the proteins are captured at the sites of their binding with DNA, ChIP helps to detect DNA-protein interactions that take place in living cells. More importantly, ChIP can be coupled to many commonly used molecular biology techniques such as PCR and real-time PCR (7), PCR with single-stranded conformational polymorphism (8), Southern blot analysis (9), Western blot analysis (10), cloning, and microarray (1–3). The resulting versatility has increased the potential of this technique.
ChIP usually involves cross-linking of the chromatin-bound proteins by formaldehyde (11–13), followed by sonication or nuclease treatment to obtain small DNA fragments (Figure 1). Immunoprecipitation is then carried out using specific antibodies to the DNA-binding protein of interest. The DNA is then released from the proteins and analyzed using various methods. ChIP has also been used to study RNA-protein interactions (14).

The steps of chromatin immunoprecipitation reaction and important alternatives used in key steps are summarized.
There are two main types of ChIP assays: X-ChIP and N-ChIP. The X-ChIP method utilizes fixed chromatin fragmented by sonication (15), while the N-ChIP uses native chromatin (16), which is unfixed and nuclease digested. The advantages and disadvantages of the two techniques are summarized in Table 1. In the N-ChIP assay, immuno-precipitation is very efficient, and the precipitated DNA can be studied without further PCR amplification (16). Antibody binding to unfixed protein is generally stronger than to the fixed protein, thus the specificity of immuno-precipitation of N-ChIP is higher than that of X-ChIP. N-ChIP is most suited to study tightly bound proteins such as histones and thus is not preferred for studying nonhistone proteins, which may get rearranged during processing. Another disadvantage of the N-ChIP protocol is that not all of the nuclease-digested chromatin is solubilized, thus a subset of chromatin remains with the nuclear pellet and is therefore excluded from the ChIP analysis. X-ChIP, on the other hand, is ideal for studying all types of proteins including nonhistone proteins. Because the X-ChIP assay is more sensitive than N-ChIP, it requires fewer cells and lesser amounts of antibody. Moreover, formaldehyde fixation reduces the possibility of protein rearrangement. The disadvantage of X-ChIP is that excess cross-linking leads to difficulty in fragmentation of the DNA to the desired size (9). This causes a smaller amount of DNA retrieval, necessitating post-ChIP amplification of the recovered DNA. There is also a risk that formaldehyde might fix transient-DNA protein interactions as well, thus interfering in the detection of stable interactions (16).
 |
Critical Parameters
Although ChIP is a highly versatile procedure, it has several limitations and requires the optimization of conditions for a successful DNA extraction. The most vital step is probably the binding of antibody, and the quality of antibody is crucial for the recovery of DNA fragments. In this section, we will discuss the critical parameters of the reaction and the alternatives available for refining the process. These parameters are summarized in Table 2.
 |
Fixation
The first step of the technique is the cross-linking of DNA and proteins. Formaldehyde is the most commonly used cross-linking agent (9). The most important advantage of using formaldehyde is the ease of reversibility of the cross-links and its ability to form bonds that span a distance of approximately 2 angstroms (17). This means that formaldehyde is able to bind molecules in close association with each other. Generally, formaldehyde is added to the medium in the cell culture flask or plate. It enters the cells through the cell membrane and cross-links the proteins to the chromatin. Formaldehyde fixation of tumor tissues and whole mouse embryos has also been done (10). In one experiment, chromatin cross-linking was carried out by the infusion of 1% formaldehyde into mouse liver in vivo, and this was followed by the dissection and mincing of the liver tissues (18). Formaldehyde causes DNA-protein, RNA-protein, as well as protein-protein cross-linking. Formaldehyde primarily targets lysine amino group and side chains of adenine, guanine, and cytosine. It is a dipolar substance and forms a Schiff's base with amino and imino groups (15). Optimal conditions for formaldehyde cross-linking need to be determined using a time-course experiment (Figure 2). Usually, nucleosomal proteins cross-link faster; longer incubation is needed for nonhistone proteins. Overfixation with formaldehyde can result in difficulty in fragmentation by sonication (Figure 2). Longer exposure to formaldehyde leads to a loss of immunoprecipitated material, although the reason for this is not understood. It is possible that prolonged cross-linking favors the binding of nucleosome-associated proteins, thereby “masking” histone epitopes. Alternatively, epitopes may be lost because of the engagement of lysines and other preferred formal-dehyde-reactive sites (15). The reaction is quenched using 0.125 M glycine (9). Reversal of cross-linking is achieved by heating at 65°C for 6 h (4).
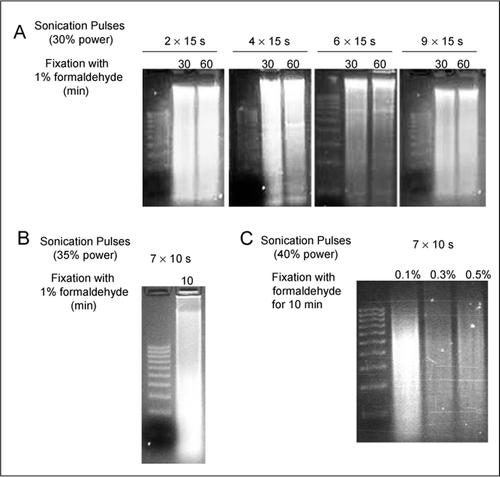
The tissue culture flasks of confluent LNCaP cells were treated with varying concentrations of formaldehyde for variable time periods. After lysis with sodium dodecyl sulfate (SDS) buffer, the chromatin was sonicated. An aliquot of sonicated chromatin was treated with proteinase K and DNA isolated and examined on an ethidium stained agarose gel. The size standards shown are a 100-bp ladder (Molecular Ruler; Bio-Rad Laboratories, Hercules, CA, USA).
Although formaldehyde is a very good agent for cross-linking, it also causes the indirect association of proteins with the DNA sequence, which could be mistaken for direct binding (4). It can also cause unfolding of protein and loss of antigen-binding sites. Moreover, following formaldehyde fixation, enzymatic digestion is not possible. Therefore, the application of other cross-linkers has been explored.
Other cross-linking agents that have been used include chemicals such as methylene blue and acridine orange (19), cisplatin (4,7,20,21), dimethylarsinic acid (22), potassium chromate (23), and ultraviolet (UV) light and lasers (24).
Chemicals
Methylene blue and acridine orange are believed to couple proteins to DNA by forming oxygen-free radicals, which diffuse away and catalyze the formation of covalent bonds between molecules that are in close contact (19,25). These methods are preferred over formaldehyde when nuclease or restriction digestion is used for chromatin fragmentation instead of sonication. Cisplatin is useful for studying nuclear matrix proteins because it fixes them more efficiently than formaldehyde (4,21,26). Unlike formaldehyde, which links proteins to DNA, RNA, or to other proteins, cisplatin cross-links proteins to DNA only, predominantly the nuclear matrix proteins and, to a lesser degree, the histone proteins. Cisplatin is used in a concentration of 0.3 to 1 mM for 30 min to 2 h at 37°C (4,27). In this case, the de-cross-linking is brought about by thiourea. However, note that cisplatin is also a very toxic substance, and one has to be careful with its use.
UV light and lasers
UV light cross-linking is performed directly on cell culture dishes kept on ice and exposed for 15 min to a source of 366 nm UV light positioned at a distance of about 2–3 cm from the dish. UV light cross-linking produces less chemical structure distortion as compared to chemical modifications (24). UV light cross-links only DNA to protein. The use of lasers has also been tried for cross-linking reactions, and several studies have been conducted. High-power lasers have been shown to increase cross-linking (24). However, both UV light and laser irradiation can damage proteins and DNA in contrast to formaldehyde, which does not damage DNA (28).
Cross-linking with more than one agent is uncommon. However, a recent report has shown that the addition of dimethyl adipimidate (DMA) as a cross-linking agent with formaldehyde helps in the binding of proteins, which could not be bound by formaldehyde alone (29). It was found that the addition of DMA produced more efficient immunoprecipitation of Rpd3 at the INO 1 promoter. DMA is an imidoester, which forms stable covalent bonds with primary amine groups and is a protein-protein cross-linking agent. However, its disadvantage is that it does not cross-link proteins to DNA and thus has to be used in combination with other cross-linkers. Examples of other serial cross-linking agents are 1-ethyl-(3-dimethylaminopropryl) carbodiimide (EDC), dimethyl pimelimidate (DMP), and disuccinimidyl glutarate (DSG) (29).
Sonication
The next important step is the sonication of the harvested chromatin. The sample is aliquoted and subjected to a number of sonication cycles. It is critical to break the DNA to 100–500 bp fragments to pinpoint the location of the DNA sequence of interest. Factors affecting sonication include the volume of the sample, depth of the sonication probe, sonication strength, and the duration of sonication (4,9). Sample volume should not exceed 1.0 mL, and the sonication depth can be increased if the sample is placed in a microfuge tube (4). The probe should be inserted to a depth of at least 1.0 cm because the chromatin solution contains sodium dodecyl sulfate (SDS), which may cause it to foam and lead to reduced sonication efficiency. Keeping the sample cool by placing it on ice also helps to reduce foaming. The output energy also needs to be adjusted to prevent foaming. Some experiments using glass beads have shown improved efficiency (9).
An alternative to sonication is nuclease digestion of the chromatin. It is used for studying N-ChIP. Note that nuclease digestion is not compatible with formaldehyde cross-linking because the cleavage is not efficient due to fixation of the chromatin (30). However, nuclease digestion is more targeted and yields fragments as small as 146 bp in size. It is performed at 37°C for 5 min. Care has to be taken during nuclease digestion because overdigestion can lead to the formation of subnucleosomal particles, thereby hindering the detection of protein-DNA interactions. Digestion can be stopped by the addition of EDTA to a final concentration of 5 mM (16). Purification of chromatin is achieved by the use of a cesium chloride (CsCl) gradient centrifugation at approximately 195, 162×g in a SW55Ti rotor (Beckman Coulter, Fullerton, CA, USA). CsCl gradient is useful in experiments involving Drosophila tissue culture cells and some human cells (31,32).
CsCl gradient helps to remove the nonchromatin proteins, lipids, and cell membrane components that may be present in the sample. Briefly, the samples are adjusted to 1.42 g/mL CsCl and added to Sarkosyl buffer (1 mM Na-EDTA, 0.5 mM Na-EGTA, 10 mM Tris-HCl, pH 8.0, adjusted to 0.5% sarkosyl) to a final volume of 5 mL. The samples are then centrifuged in a SW55Ti rotor at approximately 195,162× g for 72 h at 20°C. Fractions are collected from the bottom of the gradient using 0.25 mm capillary needles. However, this method is very time-consuming and requires 72 h of ultracentrifugation. Sonication of the fixed cells in high-detergent buffer has been used as an alternative in one study using Tetrahymena thermophila and in another using Saccharomyces cerevisiae (15,17).
Immunoprecipitation
Immunoprecipitation is the most important step in the procedure. The antibody against the protein of interest is allowed to bind to the protein-DNA complex, and the complex is then precipitated. In order to determine if the antibodies used are of ChIP-grade, the ability of the antibody to bind to the fixed material is tested by performing ChIP on formaldehyde-fixed tissues or cells (15). The activity of the antibodies on nuclear extracts in radioimmunoprecipitation (RIPA) buffer and the different detergents should be documented before performing ChIP. In addition, polyclonal antibodies are preferred over monoclonal antibodies to overcome epitope masking. To determine the efficiency of the antibody to the target antigen, Spencer et al. (4) performed immunoprecipitation using cell lysate from nonfixed cells. The optimum amount of antibody titer for maximum binding to the protein of interest should be determined empirically. Pilot experiments using increasing amounts of antibody combined with the cell lysate should be performed to establish the optimum titer. Usually the incubation is carried out overnight at 4°C with end-to-end rotation.
The immunosorbant commonly used to separate the antigen-antibody complex from the lysate is a salmon sperm DNA-protein A-Sepharose® (Upstate Biotechnology, Lake Placid, NY, USA) complex. The other immunosorbant used is protein G. Antibodies raised in rabbit have equal binding affinity for both proteins A and G, whereas antibodies raised in goat have a predilection for protein G (4). To determine the amount of protein A or G needed for the isolation of the antibody-antigen complex, the cell lysate-antibody mixture is incubated with different concentrations of the protein A or G solution, and sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) is performed. The gel is then transferred to a nitrocellulose membrane and incubated with secondary antibody and visualized. To reduce the nonspecific binding, several methods are employed. Firstly, salmon sperm DNA and bovine serum albumin (BSA) can be added to the sonicated chromatin following the incubation with primary antibody but before the addition of protein A- or Gagarose. Pretreating the protein A- or G-agarose with salmon sperm DNA and BSA and washing the beads with low-salt or high-salt buffers, lithium chloride buffer, and Tris-EDTA (TE) buffer can achieve further reduction of nonspecific binding (4). It must be noted here that the use of salmon sperm DNA as a blocking agent is not advised for ChIP-chip experiments in which nonsequence-specific amplification methods are used for analysis [e.g., ligase-mediated PCR (LM-PCR) or random-primed PCR].
A new substitute for protein A-Sepharose is protein A-coated magnetic beads. The main advantage of using the magnetic beads is that the procedure is faster when compared to the standard method. Magnetic beads may also reduce background compared to Sepharose. The main drawback of this method is the high cost. The magnetic beads are mixed with the antibody-chromatin mixture and kept on ice for 30 min with gentle mixing. The mixture is then separated using magnetic columns (16).
DNA and Protein Isolation
The immunoprecipitated DNA is eluted by incubating the beads in 250 µL 1% SDS with 0.1 M NaHCO3 for 15 min at room temperature. The eluted protein-DNA complexes are incubated at 68°C for 6 h to overnight to reverse the formaldehyde cross-links, and then at 55°C for 2 h in the presence of 200 mM NaCl, 10 mM EDTA, pH 8.0, 40 mM Tris-HCl, pH 6.5, and 50 µg/mL proteinase K to digest the protein. The sample is then extracted with a 25:24:1 mixture of phenol:chloroform:isoamyl alcohol and precipitated in the presence of carrier DNA with 2–3 volumes of absolute ethanol (4). Glycogen or tRNA is commonly used as a DNA carrier. We have obtained good results using Pellet Paint® as a DNA carrier (Novagen, San Diego, CA, USA). We prefer nonfluorescent Pellet Paint because it does not interfere with DNA quantitation when using real-time PCR (7). To remove any residual salt, the ChIP DNA is washed once with ice-cold 70% ethanol, air-dried, and resus-pended in double-distilled water or TE buffer, pH 8.0.
Proteins are isolated from the first phenol chloroform phase by the addition of BSA, 10 M H2SO4, and acetone overnight at -20°C, followed by centrifugation at 14,000× g and washing with cold acetone.
Post-Immunoprecipitation Analyses
Once the DNA of interest is isolated, many of the standard detection and quantification methods can be used to study the isolated gene fragment. The most commonly used methods include PCR, real-time PCR, slot blot hybridization, and microarray techniques.
DNA Quantitation
The DNA isolated from a ChIP assay can be analyzed by either Southern slot blot analysis or PCR (4,9). If analyzed by Southern slot blot analysis, the DNA can be applied to a nylon membrane and hybridized to radiolabeled or digoxygenin (DIG)-labeled DNA probes. Alternatively, the ChIP DNA can be radiolabeled or DIG-labeled and used as a probe to hybridize to specific genomic DNA sequences blotted on a nylon membrane (33).
The ChIP DNA quantification is commonly performed using a PCR-based approach. The PCR is carried out by using a known quantity of DNA and running identical PCRs for different numbers of cycles. The level of PCR product in each reaction is measured by agarose gel electrophoresis combined with scanning densitometry. The linear phase is determined by plotting the level of PCR products formed against the cycle number. To determine whether a specific DNA sequence becomes associated with a transcription factor or a modified histone, the levels of PCR product from ChIP and input DNA in control and treated cells are visualized by agarose gel electrophoresis using ethidium bromide to stain DNA and analyzed by scanning densitometry for quantification. The level of DNA sequence enrichment in ChIP DNA for both control and treated cells is then determined by dividing the net density of PCR product from ChIP DNA by the net density of product from the respective input DNA (4). The fold enrichment of DNA sequence in the treated sample is then determined by dividing the level of sequence enrichment for the treated sample by that for the control sample. For PCR-based detection, one crucial control is an internal loading standard. In this case, specific PCR primers are used to assay the abundance of a suspected target relative to a standard genomic fragment that is thought to be unenriched by the immunoprecipitation (such that there are four primers, two primer pairs, in each reaction). This is a critical control, without which there may be erroneous results due to the recovery of a different amount of total DNA from the ChIP and mock reactions. This internal control is especially important if the DNA from the immunoprecipitation cannot be accurately quantitated, which is often the case. Real-time PCR is based on the detection of a fluorescent signal produced during the amplification of the PCR cycle, and the real-time feature enables us to visualize the exponential part of the reaction. It basically depends on the determination of the cycle threshold (CT). CT is the cycle at which the exponential phase begins and represents the greatest rate of change in the reaction. CT is inversely proportional to the initial amount of template used. The relative quantitation of template DNA from ChIP reactions can be performed using the User Bulletin #2, ABI Prism® 7700 Sequence Detection System (Applied Biosystems, Foster City, CA, USA). The use of real-time PCR can help to identify as little as a 2-fold change in the amount of isolated DNA. The PCR product can be measured as it accumulates, and data can be collected as the reaction proceeds, increasing the sensitivity as well as precision. The results are compared to input chromatin and between the specific antibody and nonspecific immunoglobulin G (IgG; Reference 7). In our laboratory, we use real-time PCR for DNA quantitation and to calculate the ratio of bound to unbound fraction. This increases the sensitivity and reproducibility of the experiment.
Limitations of ChIP
Although the ChIP protocol is useful for studying the binding of proteins to DNA, it is usually more of a qualitative than a quantitative approach. One can determine if a specific protein is associated with a specific DNA sequence. However, whether this association is true for every cell in the cell lysate is difficult to determine. The quantification of DNA is sometimes misleading because of multiple factors, the first factor being that the cross-linking with formaldehyde is never complete (otherwise solubilization of the cross-linked nuclear DNA followed by DNA fragmentation by sonication would be impossible). From one preparation to the next, variability in cross-linking between DNA and the target protein will occur. The second factor is the immunoprecipitation step. From one experiment to the next, the antibodies may not efficiently immunoprecipitate all of the antigen. The third is the washing step. Washing the beads may result in the removal of the antibody or antigen from the beads.
In vivo Protein Biotinylation for ChIP
An effective modification of the ChIP protocol was designed by Viens et al. (34), wherein biotinylated proteins were expressed in mammalian cells in vivo. The system is based on co-expression of the target protein fused to a short biotin acceptor domain (BAD), together with the Escherichia coli biotinylating enzyme BirA. BAD is an efficient target for BirA. The open reading frame (ORF) of BirA is placed on the same vector downstream of the gene of interest. The expression of BirA from the same mRNA is ensured by internal ribosome entrance sequence (IRES) in the same vector. This provides high local concentration of the BirA enzyme, resulting in efficient biotinylation of the target protein in vivo. The technique was successfully employed to detect interactions of the transcription factor DPI with DHFR promoter in NIH/3T3 cells.
This method takes advantage of the higher avidity of the biotin-avidin interaction, thus providing greater enrichment of the target DNA. Further, it allows more stringent washing without affecting the yields, while also improving the signal-to-noise ratio. It makes the ChIP assay more robust and economical. A lesser amount of DNA is sufficient, compared to the standard assay, thus enabling scaling down of the entire reaction.
The drawback of this system is that overexpression of the biotinylated protein during transient transfection may not reflect the actual scenario in the cell with regard to the distribution of the protein in the genome. Selection of a stable cell line with low expression levels of the biotinylated protein (5%–10% of the endogenous expression levels) helps to obviate this problem (34).
ChIP in Combination With Other Methods
ChIP has been used in combination with other techniques to elucidate in vivo DNA-protein interactions. Because, in the standard ChIP procedure, specific DNA sequences are examined by PCR with gene-specific primers, one must first presume that a specific promoter might be bound by the protein of interest so as to be able to design promoter-specific primers and/or probe. This protocol, therefore, cannot be used to identify unknown gene sequences that interact with a given protein. The standard ChIP procedure has been modified to determine unknown DNA sequences that interact with a known protein without requiring prior knowledge of its target genes. ChIP cloning has been designed to isolate individual target genes. In contrast, ChIP-chip and ChIP-CpG microarray provide a more global approach (2).
ChIP Cloning
In this method, immunoprecipitated DNA from the standard ChIP method is cloned and sequenced. Several modifications were made to adapt the standard ChIP method in order to clone the immunoprecipitated DNA (2).
In the standard ChIP method, nonspecific DNA sequences are immunoprecipitated even in the absence of an antibody. In the ChIP cloning protocol, these nonspecific sequences will have an equal chance of being cloned. To decrease the amount of nonspecific DNA, two sequential immunoprecipitations are performed. However, one drawback of the second immunoprecipitation is that the amount of DNA recovered is very small. Although PCR amplification is one possible method of overcoming the low DNA yield and has been used in yeast systems (6), it may preferentially amplify some sequences but not others. To avoid this PCR bias, it has been recommended that multiple parallel immunoprecipitation reactions be performed and then pooled following the DNA purification step (2). Another important consideration in ChIP cloning is the size of the chromatin. In the standard ChIP method, it is important to sonicate the chromatin to obtain 100–500 bp fragments to localize the DNA sequence that interacts with a specific protein. In ChIP cloning, the majority of small DNA fragments represent the nonspecific precipitation of repetitive sequences, while the larger DNA fragments indicate specific binding. Therefore, it is recommended that in ChIP cloning, the chromatin should be sonicated to 1–2 kb fragments and only cloned DNA fragments of 500 bp or larger be analyzed. During sonication, DNA is sheared, causing random overhangs at the 5' and 3' ends, which makes it inefficient for cloning. In order to make the immunoprecipitated DNA competent for cloning, T4 DNA polymerase is used to create blunt ends, and the blunt-ended DNA fragments are then cloned into a vector. This method has the drawback of detecting only a small number of target genes.
ChIP-Chip
A combination of ChIP and whole genome microarrays (ChIP-chip) has been used successfully to create high-resolution genome-wide maps of in vivo DNA-protein interactions (35). The minireview by Buck and Leib (35) provides an excellent overview of the design and analysis of ChIP-chip experiments. In this method, the enriched DNA obtained by the standard procedure is labeled with a fluorescent molecule such as Cy™5 or Alexa™ 647. The input DNA is labeled with Cy3 or Alexa 555. The two probes are then combined and hybridized to a single DNA microarray. In order to obtain an unbiased survey of DNA-protein interactions, the DNA micro-arrays used in the ChIP-chip assay must contain DNA fragments that are representative of the entire genome (35). This method has been used in yeast systems to detect the binding of transcription factors to DNA over a large area in the genome. However, for this method to be successful in human systems, suitable microarrays are needed. Potential strategies to study the immediate regulatory sites include the creation of a microarray of PCR fragments representing 1 kb of DNA upstream of all known and predicted genes (2). The use of such microarrays omits interactions occurring farther than approximately 1 kb away from the arrayed element. To circumvent this problem, arrays that tile across an entire regulatory region can be designed (35). This method was employed to map the GATA-1 transcription factor to the β-globin locus (36). Arrays containing bacterial artificial chromosomes also can be used to study the distant regulatory factors (2)
ChIP-CpG Microarray
To identify a large number of genomic sites, a combination of ChIP and CpG microarray has been tried by several investigators (37,38). CpG island microarrays are preferred over cDNA microarrays if promoters are to be located. This is because the promoters are more frequently linked with CpG islands and, as a result, the possibility of isolating a transcription factor bound to the DNA is higher. Robyr and Grunstein (37) describe a method using a combination of ChIP-PCR and microarray to study histone modifications. The amplification protocol described is an adaptation of the method originally outlined by Bohlander et al. (39) and a similar method was also used by Horak and Snyder (40). Immunoprecipitated DNA from the ChIP reaction from the experimental and control strains are de-cross-linked, and PCR amplification is performed in two steps—an initial amplification by incorporation of a partially degenerate oligonucleotide, followed by a second regular PCR. Following PCR, the DNA from the experimental and the control reactions is labeled with two different fluorophores, Cy3 or Cy5. These are then hybridized to a microarray glass slide containing intergenic regions or ORFs or both. The glass slides are then scanned for both the dyes, and normalized intensities between the two strains are studied to determine the changes in acetylation patterns (37).
ChIP and In Vivo DNA Footprinting
The analysis of the interaction of proteins with either DNA or RNA sequences by in vivo footprinting involves two steps: (i) the in situ modifications of nucleic acids by the footprinting reagent and (ii) the visualization of the footprints (41). The results of DNA footprinting can be nonconclusive if the DNA sequence to be analyzed interacts with a certain protein in only a fraction of the cells (42). Furthermore, in vivo footprinting cannot indicate which protein is bound at a specific site. Lee Kang et al. (42) have combined DNA footprinting with ChIP to evolve a dynamic method of analyzing protein-DNA interactions in vivo. The method overcomes the drawbacks of the ChIP assay; namely, the failure to document the specific binding site of the protein to the DNA in vivo and the failure to distinguish in vivo protein-protein interactions from DNA-protein interactions in some cases. In this method, the chromatin is cross-linked and then nuclease digested, followed by immunoprecipitation with the appropriate antibody. The immunoprecipitated DNA-protein complex is treated with dimethylsulfate (DMS) prior to the reversal of the cross-linking. The chromatin fragments are then treated with piperidine, and the analysis is done by LM-PCR.
Concluding Remarks
The ChIP assay has become a very popular tool for studying chromatin structure and nuclear events involved in transcription. It has been used to identify target genes of many important DNA-binding proteins and their regulatory enzymes. However, it has several limitations. It is a qualitative rather than a quantitative approach. Moreover, it fails to detect simultaneous binding of multiple proteins to one target site (30). In combination with techniques such as real-time PCR, cloning, footprinting, and microarray, its use can be widened to study larger sections of the genome. New target genes and their regulatory proteins can be located. The ChIP assay needs to be adapted for tissues (35). This would enable the detection of differences in the interaction of transcription factors and promoter regions of genes in normal and neoplastic tissues, thereby shedding light on the development of neoplasia. When performed with appropriate controls, ChIP can be a very efficient approach to investigate DNA-protein interactions in vivo.
Acknowledgments
This work was supported by grants from the Department of Veterans Affairs, U.S. Army, and National Cancer Institute (to R.S.). R.S. is a recipient of the “Advanced Research Cancer Development Award” from the Department of Veterans Affairs.
Competing Interests Statement
The authors declare no competing interests.
References
- 1. . 2001. Use of chromatin immunoprecipitation to clone novel E2F target promoters. Mol. Cell Biol. 21:6820–6832.
- 2. . 2002. Identification of unknown target genes of human transcription factors using chromatin immunoprecipitation. Methods 26:37–47.
- 3. . 2002. Isolating human transcription factor targets by coupling chromatin immunoprecipitation and CpG island microarray analysis. Genes Dev. 16:235–244.
- 4. . 2003. Chromatin immunoprecipitation: a tool for studying histone acetylation and transcription factor binding. Methods 31:67–75.
- 5. . 2003. Analysis of specific lysine histone H3 and H4 acetylation and methylation status in clones of cells with a gene silenced by nickel exposure. Toxicol. Appl. Pharmacol. 190:272–277.
- 6. . 2000. Genome-wide location and function of DNA binding proteins. Science 290:2306–2309.
- 7. 2002. Methylation of alpha-type embryonic globin gene alpha pi represses transcription in primary erythroid cells. Blood 100:4217–4222.
- 8. . 1999. Analysis of chromatin in limited numbers of cells: a PCR-SSCP based assay of allele-specific nuclease sensitivity. Nucleic Acids Res. 27:e32.
- 9. . 1997. Analysis of chromatin structure by in vivo formaldehyde cross-linking. Methods 11:205–214.
- 10. . 2002. Characterizing transcription factor binding sites using formaldehyde crosslinking and immunoprecipitation. Methods 26:48–56.
- 11. . 1981. Use of whole-cell fixation to visualize replicating and maturing simian virus 40: identification of new viral gene product. Proc. Natl. Acad. Sci. USA 78:6081–6085.
- 12. . 1999. In vivo cross-linking and immunoprecipitation for studying dynamic protein:DNA associations in a chromatin environment. Methods 19:425–433.
- 13. . 1981. A new method for the isolation of replicative chromatin: selective deposition of histone on both new and old DNA. Cell 23:121–134.
- 14. . 2002. Reversible cross-linking combined with immunoprecipitation to study RNA-protein interactions in vivo. Methods 26:182–190.
- 15. 2000. Mapping chromosomal proteins in vivo by formaldehyde-crosslinked-chromatin immunoprecipitation. Trends Biochem. Sci. 25:99–104.
- 16. . 2003. Immunoprecipitation of native chromatin: NChIP. Methods 31:76–82.
- 17. . 1991. A simplified formaldehyde fixation and immunoprecipitation technique for studying protein-DNA interactions. Anal. Biochem. 197:83–90.
- 18. . 2001. Transcription factor FoxA (HNF3) on a nucleosome at an enhancer complex in liver chromatin. J. Biol. Chem. 276:44385–44389.
- 19. . 2000. The specificity of protein-DNA crosslinking by form-aldehyde: in vitro and in drosophila embryos. Nucleic Acids Res. 28:e4.
- 20. 1995. The nuclear matrix and the regulation of chromatin organization and function. Int. Rev. Cytol. 162A:191–250.
- 21. . 2001. Altered profiles in nuclear matrix proteins associated with DNA in situ during progression of breast cancer cells. Cancer Res. 61:1362–1366.
- 22. . 1993. Crosslink formation between DNA and nuclear proteins by in vivo and in vitro exposure of cells to dimethylarsinic acid. Biochem. Biophys. Res. Commun. 191:1184–1191.
- 23. . 1991. Complexing of actin and other nuclear proteins to DNA by cis-diamminedichloroplatinum (II) and chromium compounds. Carcinogenesis 12:269–276.
- 24. . 1999. Crosslinking of proteins to DNA in human nuclei using a 60 femtosecond 266 nm laser. Nucleic Acids Res. 27:3676–3684.
- 25. . 1993. Photochemical interactions of methylene blue and analogues with DNA and other biological substrates. J. Photochem. Photobiol. B 21:103–124.
- 26. . 1979. Binding of cis- and trans-dichlorodiammineplatinum (II) to the nucleosome core. Proc. Natl. Acad. Sci. USA 76:6091–6095.
- 27. . 1998. In situ cross-linking by cisplatin of nuclear matrix-bound transcription factors to nuclear DNA of human breast cancer cells. Cancer Res. 58:3004–3008.
- 28. . 1976. On the use of ultraviolet light to study protein-DNA crosslinking. Nucleic Acids Res. 3:1065–1071.
- 29. . 2003. In vivo protein-protein and protein-DNA cross-linking for genomewide binding microarray. Methods 31:90–95.
- 30. . 2002. Dissecting long-range transcriptional mechanisms by chromatin immunoprecipitation. Methods 26:27–36.
- 31. . 1998. New POU dimer configuration mediates antagonistic control of an osteopontin preimplantation enhancer by Oct-4 and Sox-2. Genes Dev. 12:2073–2090.
- 32. . 1999. Virus infection leads to localized hyperacetylation of histones H3 and H4 at the IFN-beta promoter. Mol. Cell 3:125–129.
- 33. . 1999. Increased Ser-10 phosphorylation of histone H3 in mitogen-stimulated and oncogene-transformed mouse fibro-blasts. J. Biol. Chem. 274:24914–24920.
- 34. . 2004. Use of protein biotinylation in vivo for chromatin immunoprecipitation. Anal. Biochem. 325:68–76.
- 35. . 2004. ChIP-chip: considerations for the design, analysis, and application of genome-wide chromatin immuno-precipitation experiments. Genomics 83:349–360.
- 36. . 2002. GATA-1 binding sites mapped in the beta-globin locus by using mammalian chIp-chip analysis. Proc. Natl. Acad. Sci. USA 99:2924–2929.
- 37. . 2003. Genome-wide histone acetylation microarrays. Methods 31:83–89.
- 38. . 2001. Genomic binding sites of the yeast cell-cycle transcription factors SBF and MBF. Nature 409:533–538.
- 39. . 1992. A method for the rapid sequence-independent amplification of microdissected chromosomal material. Genomics 13:1322–1324.
- 40. . 2002. ChIP-chip: a genomic approach for identifying transcription factor binding sites. Methods Enzymol 350:469–483.
- 41. . 1997. In vivo footprinting of the interaction of proteins with DNA and RNA. Methods 11:151–163.
- 42. . 2002. Combining chromatin immunoprecipitation and DNA footprinting: a novel method to analyze protein-DNA interactions in vivo. Nucleic Acids Res. 30:e44.



