








Useful tool to generate unidirectional deletion vectors by utilizing the star activity of BamHI in an NcoI-BamHI-XhoI cassette
It has been shown that restriction endonucleases can cleave DNA sequences that are similar but not identical to their cognate recognition sequences under some nonstandard conditions (1). This phenomenon is mediated by the so-called “star” activity of restriction endonucleases. In general, biologists try to avoid star activity when performing restriction endonuclease digests in order to achieve specific cleavage of DNAs. However, we have found a way to take advantage of this adverse property of restriction endonucleases to facilitate molecular cloning. In this study, we developed a rapid, convenient, economical, and versatile method (diagrammed in Figure 1A) to facilitate large-scale molecular cloning and functional analyses of gene products.
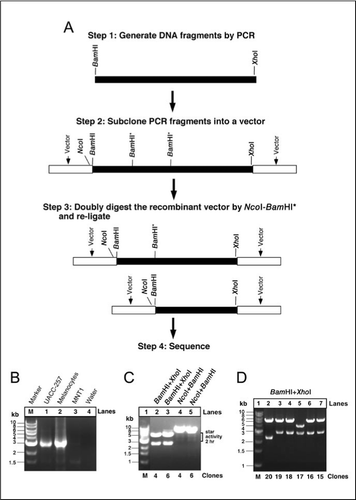
(A) Presentation of the steps for generating deletion constructs by utilization of the “star” activity of endonucleases. Step 1: generate DNA fragments with desired endonuclease restiction sites (e.g., BamHI-DNA-XhoI) by PCR; step 2: subclone desired fragments into a desired vector containing the restriction sites of interest (e.g., the NcoI site in pTM1); step 3: doubly digest a vector of interest (e.g., pTM1/NcoI-BamHI-DNA-XhoI) by enzymes with one having known star activity (e.g., BamHI), examine the digestion pattern from 2-24 h, excise digested bands (those containing smears), and self-ligate them by T4 DNA ligase; step 4: mini-prepare plasmids and doubly digest by BamHI/XhoI to verify the size of deletions and confirm the results by directly sequencing plasmids of interest. The vector is presented as a linear fragment. (B) Reverse transcription PCR (RT-PCR) cloning ABCB5: cDNAs from human melanocytes and melanoma cell lines (UACC-257 and MNT-1) were subjected to 35 cycles of PCR using the primer set of ABCB5 as described in the text. (C) Subcloning of ABCB5 into the pTM1 vector: the 2.9-kb fragments derived from RT-PCR were doubly digested with BamHI and XhoI and ligated into the BamHI and XhoI sites of pTM1. The plasmids from the recombinant clones 4 and 6 were doubly digested by BamHI-XhoI. Both clones 4 and 6 were also subjected to NcoI-BamHI digest at 37°C for either 2 h or 24 h (data not shown). The bands that include 7.8-8.3 kb of both lanes 4 and 5 were excised, the gel was purified using the GENECLEAN II Kit, self-ligated by DNA T4 ligase, and transformed into the Escherichia coli cells. (D) The plasmids from the ligated clones were subjected to BamHI-XhoI digest to verify the length of the inserts. Both the PCR products and plasmid digests were analyzed by 1% agarose gel and stained with ethidium bromide. M, molecular weight marker (1-kb DNA Ladder; New England Biolabs).
In this report, we use the human ABCB5 gene, a novel ATP-binding cassette (ABC) transporter, as our target gene. The ABCB5-specific primers used were the ABCB5 forward primer (5′-AAGGATCCATTGCTTCTCGGCCTTTTGGCTAAG-3′) and the reverse primer (5′-AACTCGAGTCAC-TGCACTGACTGTGCATTCACT-3′) (K.G. Chen and M.M. Gottesman, unpublished data). The boldfaced and underlined bases represent the restriction sites BamHI and XhoI, which we used to facilitate subcloning and exploit the star activity of BamHI. All restriction endonucleases used in this study were purchased from New England Biolabs (Beverly, MA, USA). A conventional reverse transcription PCR (RT-PCR) protocol was used to amplify the ABCB5 cDNA sequences. The total cDNAs were subjected to denaturation at 94°C for 30 s, followed by 35 cycles of amplification for 30 s at 94°C, 30 s at 55°C, and 3 min at 72°C.
In order to generate unidirectional star activity of an enzyme in an expression cassette, the PCR fragments were doubly digested by both BamHI and XhoI in the presence of SuRE/Cut Buffer B (10 mM Tris-HCl, 5 mM MgCl2, 100 mM NaCl, 1 mM 2-mercaptoethanol, pH 8.0, at 37°C) from Roche Applied Science (Indianapolis, IN, USA) and then subcloned into the BamHI and XhoI sites of the pTM1 vector, a vaccinia virus-based expression vector that has been extensively used in our laboratory for membrane protein expression (2). To generate serial deletion constructs for pTM1 so that the first ATG site of ABCB5 for the protein translation was placed closely downstream from the NcoI site to achieve high transcriptional efficiency (2,3), we further doubly digested the pTM1-ABCB5 (NcoI-BamHI-XhoI) vector by NcoI-BamHI in the presence of both NEBuffer 2 [10 mM Tris-HCl, 10 mM MgCl2, 50 mM NaCl, 1 mM dithiothreitol (DTT), pH 7.9] and bovine serum albumin (BSA; 100 µg/mL) at 37°C for 2 h or 24 h, which is likely to produce a digestion pattern caused by the star activity of the enzymes. The digested sample was subjected to electrophoresis on a 2% agarose gel, and the desired fragments were excised, purified by the GENECLEAN® II Kit (Qbiogene, Carlsbad, CA, USA), and directly ligated by DNA T4 ligase (Roche Applied Science) at 16°C overnight. Two microliters of the ligates were used to transform the Escherichia coli (DH5α) strain (Invitrogen, La Jolla, CA, USA). The plasmids with altered sizes were isolated and sequenced using the sequencing core facility at the National Cancer Institute (Bethesda, MD, USA).
The ABCB5 PCR fragments (approximately 2.9 kb, Figure 1B) were obtained by PCR and subcloned into the BamHI and XhoI sites of the pTM1 vector (Figure 2A) to obtain pTM1-ABCB5 (NcoI-BamI-XhoI) (Figure 2B). We further doubly digested the pTM1-ABCB5 (NcoI-BamHI-XhoI) vector by NcoI-BamHI, which produced a digestion pattern caused by the star activity of the enzymes (Figure 1C). The plasmids with altered sizes were identified (Figure 1D). Of these, the majority of clones contained an intact backbone of the pTM1 vector, as indicated by a 5.4-kb band. The length of the inserts (the ABCB5 fragments) identifies the fragments that were cleaved by BamHI or BamHI* (BamHI star activity). For example, one clone contained a 2.4-kb insert that was the result of a deletion of a large fragment of ABCB5 (Figure 1D, clone 20). This has been confirmed by sequencing (Figure 2C).

(A) Presentation of the structure of the pTM1 vector and its multiple cloning sites (MCS), which is based on the vaccinia virus-bacteriophage T7 RNA polymerase system (2,3). (B) The structure of the pTM1-ABCB5 (NcoI-BamHI-XhoI) cassette. (C) Generation of the deletion construct of ABCB5 by NcoI-BamHI digest: the breakpoint and rejoining sites were indicated and sequenced. NcoI* denotes the altered NcoI site (from CCATGG to CATATGG), possibly due to an altered digestion and repair. All plasmids are represented as linear fragments by splitting the ampicillin resistance gene (Amp).
It is believed that any restriction endonuclease can be made to cleave noncanonical sites under certain altered conditions (such as changes in pH or ionic strength; References 4–6). New England Biolabs has confirmed that many restriction endonucleases exhibit star activity. We have noted that star activity was correlated with the length of the enzyme digestion (more than 2 h) in a water bath due to an altered ionic strength resulting from the evaporation of water from the reaction mixture in the Eppendorf® tube, which condenses at the top of the tube. The most common types of altered activity can result in single base substitutions, disruption of the outer bases in a cognate sequence, and single-strand nicking (4). Previous studies showed that EcoRI* cut the sequence NAATTN or any sites that differ in a single base change under certain conditions, such as an elevated pH and low ionic strength (5,6). We designed an NcoI-BamHI-XhoI cassette in the pTM1 vector, in which our gene of interest was inserted into the BamHI-XhoI sites, resulting in the NcoI-BamHI-ABCB5 (2.9 kb)-XhoI cassette (Figure 2B). The star activity of NcoI was limited in this NcoI-BamHI combination after the linearization of the plasmids. Moreover, the star activities of NcoI and BamHI are mutually exclusive in this combined digest, which allows for unidirectional generation of deletions (Figure 1D, clones 20 and 17). It allows BamHI to exert its star activity in a unidirectional manner, whereas NcoI* might produce a nick in a DNA strand (Figure 2C). Thus, BamHI, which recognizes and cleaves the motif sequence GGATCC, displayed a new phenomenon that involves the relaxation of sequence specificity and cleaves a noncognate site, TGATCC, under a prolonged reaction in the presence of NcoI. In this study, we chose BamHI* simply as one example to demonstrate the feasibility of our proposed application. The restriction endonucleases from New England Biolabs that have been demonstrated to exhibit star activity in the cassette shown in Figure 2A include BamHI, EcoRI, PstI, and SalI. Other restriction endonucleases include ApoI, AseI, BssHII, EcoRV, HindIII, HinfI, PvuII, ScaI, TaqI, and XmnI (see New England Biolabs, http://www.neb.com/neb). Thus, the redundancy of the above restriction endonucleases with star activity would allow us to choose different enzyme combinations for a given gene sequence to generate the desired constructs. Our procedures for generating a cluster of constructs can be completed in approximately 4 days, compared to other conventional techniques, which usually take about 1 week to produce the desired constructs. Clearly, this technique provides a rapid, convenient, and economical way to generate unidirectional deletions.
Acknowledgments
We would like to thank George Leiman for assistance in the preparation of the manuscript and figures.
Competing Interests Statement
The authors declare no competing interests.
References
- 1. . 1986. Relaxation of recognition sequence of specific endonuclease HindIII. Nucleic Acids Res. 14:811–821.
- 2. . 1998. Functional expression of human P-glycoprotein from plasmids using vaccinia virus-bacteriophage T7 RNA polymerase system. Methods Enzymol. 292:456–473.
- 3. . 1991. Propagation of cell cultures and vaccinia virus stocks, 16.16.1–16.16.7. In F.M. Ausubel, R. Brent, R.E. Kinston, D.D. Moore, J.A. Smith, J.C. Seidman, and K. Struhl (Eds.), Current Protocols in Molecular Biology. John Wiley and Sons, New York.
- 4. 1988. The TaqI “star” reaction: strand preferences reveal hydrogen-bond donor and acceptor sites in canonical sequence recognition. Gene 65:149–165.
- 5. . 1975. Specificity of substrate recognition by the EcoRI restriction endonuclease. Proc. Natl. Acad. Sci. USA 72:3310–3314.
- 6. . 1982. Cloning and sequencing of restriction fragments generated by EcoRI*. DNA 1:109–115.



