








Improved yield and accuracy for DNA extraction in microbiome studies with variation in microbial biomass
Abstract
A major challenge for microbiome studies is maintaining an even and accurate DNA extraction in the presence of samples with a wide range of bacterial content. Here we compare five DNA extraction methods using replicate stool samples that were diluted to create high and low biomass samples. Our results indicate greater variation in microbiome composition between high and low biomass samples than variation between methods. Many of the extraction methods had reduced yield from low biomass samples; however, our adapted plate column-based extraction method was evenly efficient and captured the largest number of high-quality reads. Based on these results, we have identified a DNA extraction method that ensures adequate yield in metagenomic microbiome studies that have samples with a broad range of bacterial content.
METHOD SUMMARY
A high-throughput DNA extraction method for studies that have samples with a broad range of bacterial content for 16S rRNA gene amplicon and shotgun metagenomic sequencing analyses. This consists of enzyme pretreatment, followed by adapting a plate column-based protocol to increase yield from low biomass samples.
A major barrier for increasing the reproducibility and accuracy of microbiome amplicon-based sequencing is efficient DNA extraction from samples that differ in bacterial biomass content. In clinical microbiome research, this is often encountered in patients that have severely disturbed microbiomes or when comparing samples from multiple body sites [1,2]. This is also commonly encountered in environmental microbiome research as samples can vary widely in their bacterial content [3–5].
Multiple steps in microbiome pipelines can introduce technical errors, including: sample processing, DNA extraction, PCR amplification of 16S rRNA gene, and sequencing and classification. Improvements have been made in reducing errors and biases from sample storage, differential cell lysis due to differences in cell wall composition, amplification of reagent contaminants, chimera generation, inadequate coverage by 16S rRNA primers, PCR and sequencing errors [6–9]. Nevertheless, DNA extraction continues to be a large source of experimental variability in 16S rRNA sequence analysis and the most time-consuming step in a high-throughput pipeline [6]. Although DNA extraction protocols have been published for low bacterial biomasses [10,11], they are hard to implement as part of a high-throughput pipeline. Further, those focused on low biomass can become saturated with high biomass samples leading to uneven sequence sampling bias. This is problematic for studies analyzing samples with a wide range of bacterial biomass. For example, studies of hematopoietic stem cell transplant (HSCT) recipients often contain participants who have undergone multiple courses of antibiotic treatments, chemotherapy, episodes of diarrhea and other disruptions of their microbiomes. In previous stool microbiome studies of HSCT recipients, 13–65% of samples were excluded due to low bacterial burden [12–14].
We aimed to identify a DNA extraction method that yields adequate DNA for studies with samples having a large range of bacterial biomass while preserving taxonomic accuracy. We compared a modified column-based extraction protocol with commercially available column-based and magnetic bead-based protocols.
To compare the extraction efficiency and ability to eliminate PCR inhibitors, we used deidentified stool from 14 healthy children and 79 HSCT recipients collected with St Jude IRB approval. To assess the full range of extraction efficiency with different extraction methods, we prepared replicates from aliquots of healthy children's samples and extracted each sample separately. A dilution curve from a single healthy child was used to determine linearity of extraction efficiency. For positive control, we used a defined mock community ZymoBIOMICS Microbial Community Standard (Zymo Research, CA, USA). For negative control, we used phosphate-buffered saline (PBS).
We adapted the QIAamp BiOstic Bacteremia DNA Kit (Qiagen, Hilden, Germany), which had been optimized for low bacterial content samples for use with a 96-well silica membrane column (from DNEASY POWERSOIL HTP Kit [Qiagen]. This replacement (referred to as SJ) significantly reduces sample preparation time for high-throughput studies compared to single tube QIAamp BiOstic Bacteremia DNA Kit (Figure 1C). The modified, custom kit developed in this study can be ordered as the DNeasy PowerSoil 96 HTP (384; Cat. No. 1115096). Starting with 200 mg of stool from a single healthy child, dilutions from 10% w/v (20 mg) to 0.00001% w/v (20 ng) were prepared in PBS [15]. These replicates were extracted in parallel to compare the yield of standard column-based and magnetic bead-based nucleic acid purification protocols: FastDNA-96™ Soil Microbe DNA kit (MP Biomedicals, Solon, OH) (MP), DNEASY POWERSOIL HTP Kit (PS), QIAamp BiOstic Bacteremia DNA Kit (Qiagen) (BA) and MagAttract PowerSoil DNA KF Kit (KF) using KingFisher Flex Purification System (ThermoFisher Scientific, MA, USA). In addition, replicate 10% w/v (20 mg) of the 14 healthy children were extracted seperately with the four methods (SJ, MP, BA, KF) to compare extraction efficiency and compositional changes in high biomass samples. Extractions were performed according to manufacturer's protocol. Prior to extraction, all samples were incubated for 1 h at 37°C with enzyme combination 50 μl of Lysozyme (10 mg/ml), 6 μl Mutanolysin (25 kU/ml), 3 μl Lyostaphin (4 KU/ml), except for MP. Total DNA content was assessed using Qubit dsDNA HS Assay Kit (Invitrogen, CA, USA), 10 μl of the extraction was assayed as a comparative method to determine the total amount of DNA present in the sample, including human and other microbial DNA. Bacterial DNA content was assessed using a 16S rRNA quantitative PCR using a plasmid containing E. coli 16S gene as the standard, primers and probes as previously described [16]. Values from the 16S rRNA quantitative PCR were used to standardize the DNA quantity input for 16S rRNA gene amplicon sequencing as previously described [16].

DNA extracted by the SJ and BA methods maintains linearity of extraction efficiency across 10% to 1E-7% w/v dilution of stool (A). Dotted line is expected yield of bacterial DNA content for each method inferred from dilutions and 10% w/v stool yield. All methods showed discrepancy in number of quality filtered reads from sequenced DNA extracted from high and low biomass stool (B) and differences in the average time needed to process 96 samples (C).
BA: QIAamp BiOstic Bacteremia DNA Kit; KF: MagAttract PowerSoil DNA KF Kit; MP: FastDNA-96™ Soil Microbe DNA kit; PS: DNEASY POWERSOIL HTP Kit; SJ: DNEASY POWERSOIL HTP Kit.
The BA and SJ methods had the most consistent high yield of bacterial DNA content across the dilutions, with multiple kits failing to adequately capture bacterial DNA to maintain linearity at lower dilutions (Figure 1A). To assess how extraction yield impacts ability to obtain representative communities for high and low biomass samples we evaluated the number of reads obtained from 16S rRNA gene amplicon sequencing (paired end 2 × 300bp on miseq of V3–V4 region of 16S). The SJ method had the least discrepancy in number of quality reads (as assessed after quality filtering with DADA2 [17]) between high biomass and diluted stool (Figure 1B). The BA kit is designed for extraction of highly inhibited, low bacterial biomass samples and surprisingly had a higher yield of quality reads for the lower biomass sample (maybe due to saturation with the higher biomass sample). As a single tube protocol, the processing time makes the BA kit impractical for studies with large sample numbers (Figure 1C). As expected, all of the methods had adequate yields for samples with high bacterial biomass content in terms of total DNA content, bacterial DNA content and quality filtered reads (Figure 2A–C).

Total DNA yield (A) and bacterial DNA content (B) for high biomass samples were similar for all methods, except MP which was statistically different by non-parametric binomial two-sided sign test (*<0.05; **<0.01, ***<0.001). All methods yielded similar average number of quality filtered reads (C) when DNA extracted was sequenced (error bars represent SEM).
BA: QIAamp BiOstic Bacteremia DNA Kit; KF: MagAttract PowerSoil DNA KF Kit; MP: FastDNA-96™ Soil Microbe DNA kit; PS: DNEASY POWERSOIL HTP Kit; SJ: DNEASY POWERSOIL HTP Kit.
To ensure that the increased yield seen with the adapted extraction method (SJ) did not allow for increased retention of PCR inhibitory substances we assessed using an internal amplification control (IAC) as previously described by others [18]. We found no evidence for PCR inhibition in any of the extraction methods tested.
To assess the bias introduced by extraction method we prepared DNA extracted with different kits from 14 healthy children, positive mock community and negative control for 16S rRNA gene amplicon sequencing as previously described by others [16]. Paired-end reads were quality filtered and classified in DADA2 using the RDP version 16 (March 2018) [17]. The accuracy of the positive mock microbial community was quantified as the root mean square deviation between observations and expectations. Figure 3A and 3B show that the extraction methods differed notably across different genera in their overall accuracy. The overall bias introduced by extraction methods was equivalent for mock community between methods (no significant difference; paired t test). The inter-sample variation among 14 healthy children (median across samples; Bray–Curtis Distance was 0.57) was greater than the bias introduced by extraction method (Bray–Curtis Distance 0.27), seen in Figure 3C. However, there was a great deal of variation within an extraction method between a high biomass and low biomass (diluted) replicate of the same sample (Bray–Curtis Distance 0.43). Taken together, these results show that discrepancies in bacterial biomass of samples can result in substantial technical variation, highlighting the importance of choosing the proper DNA extraction method.

Extraction of mock community showed differences from expected (dotted line) relative abundance per each genus (A) and per total community as root mean square deviation (B). Relative effect size of different sources of variation in sample composition of high and low biomass samples as measured by Bray-Curtis distance (C) and pairwise comparison between methods show which are comparable (D).
BA: QIAamp BiOstic Bacteremia DNA Kit; KF: MagAttract PowerSoil DNA KF Kit; MP: FastDNA-96™ Soil Microbe DNA kit; PS: DNEASY POWERSOIL HTP Kit; SJ: DNEASY POWERSOIL HTP Kit.
To determine if there was any compositional influence on extraction efficiency, we compared relative abundance of each taxa in one method to the relative abundances of that taxa in all other methods with a Kruskal–Wallis test. There were no differences seen between the extraction methods in terms of taxa-specific abundances (p-values corrected for false discovery rate were all >0.05). This may be due to the small number of methods compared or possibly increased efficiency of gram-positive bacteria DNA extraction in these particular methods (with both bead-beating and enzyme lysis steps).
Lastly, in order to validate that the increased yield seen in the SJ modified protocol allowed for a greater number of samples to be maintained in a HSCT cohort, we compared the outcome of this kit to that of the MP Soil extraction method. Two samples from each participant in the cohort were extracted with either the SJ or MP kits; sequencing and sequence processing was as outlined above. The number of usable high-quality sequencing reads from the two kits was compared, as shown in Figure 4. The number of usable sequencing reads was higher with the SJ modified protocol, even in patients that had lower bacterial content stool.
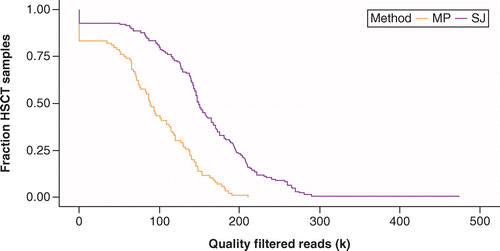
Kaplan–Meir curve showing fraction of HSCT samples versus number of quality filtered reads for SJ and MP methods.
HSCT: Hematopoietic stem cell transplant; MP: FastDNA-96™ Soil Microbe DNA kit; SJ: DNEASY POWERSOIL HTP Kit.
In conclusion, we have modified a column-based plate extraction kit for comparison to other kits in terms of reliable extraction of DNA in low bacterial biomass samples for 16S rRNA sequencing results. The reliability will inherently be lower in low bacterial biomass sampling due to amplification biases and under sampling, but efficient extraction can mitigate some of these effects [19,20]. Ultimately, we found that the modified protocol is efficient for low bacterial biomass samples, allowed for more high-quality amplicon sequences to be produced, did not take significantly more time, and provided comparable community compositions. By increasing DNA yields, we can more accurately focus on minute changes in the gut microbiome of HSCT patients without having to exclude samples for low bacterial content. This modified high-throughput extraction method will allow for more accurate microbiome composition in environmental and clinical studies where samples vary widely in bacterial content.
Author contributions
EM conceived the idea for the study. RH and GM organized and processed sample collection. AD and CK performed the experiments. EM, AD, CK, AR analyzed the data and drafted the manuscript. All authors approved the final manuscript prior to submission.
Acknowledgments
The authors thank the Hartwell Center for Biotechnology for their services and Ronald Dallas for help organizing HSCT sample collection.
Financial & competing interests disclosure
This work was funded by ALSAC. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
Ethical conduct of research
The authors state that they have obtained appropriate institutional review board approval and informed consent has been obtained from the participants involved.
Open access
This work is licensed under the Attribution-NonCommercial-NoDerivatives 4.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
Supplementary data
To view the supplementary data that accompany this paper please visit the journal website at: www.future-science.com/doi/suppl/10.2144/btn-2019-0014
References
- 1. Identification of low abundance microbiome in clinical samples using whole genome sequencing. Genome Biol. 16, 265 (2015).
- 2. The effects of airway microbiome on corticosteroid responsiveness in asthma. Am. J. Respir. Crit. Care Med. 188(10), 1193–1201 (2013).
- 3. New perspectives on viable microbial communities in low-biomass cleanroom environments. ISME J. 7(2), 312–324 (2013).
- 4. . Changes in the drinking water microbiome: effects of water treatments along the flow of two drinking water treatment plants in a urbanized area, Milan (Italy). Front. Microbiol. 9, 2557 (2018).
- 5. . DNA extraction from low-biomass carbonate rock: an improved method with reduced contamination and the low-biomass contaminant database. J. Microbiol. Methods 66(1), 21–31 (2006).
- 6. . Evaluation of methods for the extraction and purification of DNA from the human microbiome. PloS One 7(3), e33865 (2012).
- 7. Sampling and pyrosequencing methods for characterizing bacterial communities in the human gut using 16S sequence tags. BMC Microbiol. 10, 206 (2010).
- 8. The impact of different DNA extraction kits and laboratories upon the assessment of human gut microbiota composition by 16S rRNA gene sequencing. PloS One 9(2), e88982 (2014).
- 9. . DNA extraction for streamlined metagenomics of diverse environmental samples. Biotechniques 62(6), 290–293 (2017).
- 10. . Protocol improvements for low concentration DNA-based bioaerosol sampling and analysis. PloS One 10(11), e0141158 (2015).
- 11. An Improved Method for High Quality Metagenomics DNA Extraction from Human and Environmental Samples. Sci. Rep. 6, 26775 (2016).
- 12. Enteric microbiome markers as early predictors of clinical outcome in allogeneic hematopoietic stem cell transplant: results of a prospective study in adult patients. Open Forum Infectious Dis. 4(4), ofx215 (2017).
- 13. Antibiotic-induced depletion of anti-inflammatory Clostridia is associated with the development of graft-versus-host disease in pediatric stem cell transplantation patients. J. Am. Soc. Blood Marrow Transplant. 23(5), 820–829 (2017).
- 14. Metagenomic analysis of the stool microbiome in patients receiving allogeneic stem cell transplantation: loss of diversity is associated with use of systemic antibiotics and more pronounced in gastrointestinal graft-versus-host disease. J. Am. Soc. Blood Marrow Transplant. 20(5), 640–645 (2014).
- 15. . Impact of DNA extraction, sample dilution, and reagent contamination on 16S rRNA gene sequencing of human feces. Appl. Microbiol. Biotechnol. 102(1), 403–411 (2018).
- 16. Stool microbiota at neutrophil recovery is predictive for severe acute graft vs host disease after hematopoietic cell transplantation. Clin. Infect. Dis. 65(12), 1984–1991 (2017).
- 17. . DADA2: high-resolution sample inference from Illumina amplicon data. Nat. Methods 13(7), 581–583 (2016).
- 18. . Development and optimization of quantitative PCR for the diagnosis of invasive aspergillosis with bronchoalveolar lavage fluid. BMC Infect. Dis. 8, 73 (2008).
- 19. Systematic improvement of amplicon marker gene methods for increased accuracy in microbiome studies. Nat. Biotechnol. 34(9), 942–949 (2016).
- 20. Random sampling process leads to overestimation of beta-diversity of microbial communities. MBio. 4(3), e00324–00313 (2013).



