







Development and validation of cell-based assays for the detection of neutralizing antibodies to drug products: a practical approach
Abstract
Neutralizing antibodies (NAbs) that bind to drug products and may diminish or eliminate the associated biological activity are an unintended and undesirable outcome of some drug products. Standard immunoassays can detect drug-specific antibodies but cannot distinguish NAbs, so cell-based assays are often preferred because they closely mimic the mechanism by which NAbs and drug products interact in vivo. Each cell-based NAb assay is unique and based on several factors, such as the drug product, study population and phase of development (preclinical or clinical). The type of NAb assay (direct or indirect) depends on the drug’s mechanism of action. Key steps in assay development are: selecting a suitable cell line, choosing the proper cellular response (end point method), selection of proper controls and optimization of assay parameters. Once developed, the assay must be rigorously tested (validated) to ensure that it meets several important criteria and is fit for its intended purpose.
NAb: Neutralizing antibody.

NAb: Neutralizing antibody.

NAb: Neutralizing antibody.


Many drug products currently under development, including growth factors, monoclonal antibodies, cytokines and their biosimilars, are large therapeutic molecules that carry the risk of provoking an unwanted immune response in the patient [1–6]. Factors that may increase immunogenicity include size and purity of the protein, manufacturing process, patient health status and clinical indication, among others [7–10]. This immunogenicity, which may result in the formation of antidrug antibodies, can lead to a range of clinical outcomes (from no adverse effect to severe harm) due to altered efficacy, safety or PKs of the drug product. Neutralizing antibodies (NAbs), a subset of antidrug antibodies, bind to the drug product and may diminish or eliminate its biological activity [4,5]. As an example, one recent study found neutralizing anti-IFN-γ autoantibodies in 88% of Asian adults with multiple opportunistic infections. These autoantibodies were associated with an adult-onset immunodeficiency akin to that of advanced HIV infection [11].
NAbs may also cross-react with the endogenous analyte in addition to the drug product. One well-known example of cross-reactivity involved erythropoietin (EPO), a hormone that regulates red blood cell production. Patients treated with recombinant human EPO for anemia of chronic renal failure initially responded to the drug, but then developed severe transfusion-dependent anemia due to pure red cell aplasia. A detailed study of these patients and others with similar reactions demonstrated the presence of NAbs that bound to both recombinant and endogenous EPO, completely disrupting red blood cell production [12,13].
As a result, the US FDA, the European Medicines Agency and other regulatory bodies have issued similar guidance documents regarding the development of valid, sensitive immunoassays and cell-based assays as part of the drug-development process [14,15]. While standard immunoassays, such as enzyme-linked immunosorbent assays (ELISA) and radioimmunoprecipitation assays, can detect antidrug antibodies, these tests cannot discriminate how these antibodies interact with the drug product. Detecting NAbs requires the use of more specialized, in vitro, mammalian-cell-based assays or non-cell-based competitive ligand-binding assays. While both types of assay are useful, many experts recommend the cell-based assay platform as it “most closely mimics the mechanism by which NAbs may exert their effect in vivo”[7].
Each NAb assay is unique and depends on the drug product (e.g., monoclonal antibody, protein, oligosaccharide or soluble receptor), the study population (normal or diseased) and the phase of study (preclinical or clinical). The type of NAb assay (direct or indirect) depends on the drug’s mechanism of action. Most NAb assays are performed in a 96-well plate, but can be adapted for high-throughput and automation if needed. Key steps in designing and testing a cell-based NAb assay include:
▪ Selecting a suitable cell line;
▪ Choosing the proper cellular response (end point method);
▪ Selection of proper controls;
▪ Optimization of assay parameters;
▪ Validation.
The purpose of this paper is to provide practical guidance on the development and validation of cell-based assays for the detection of NAbs to therapeutic proteins. The recommendations are based on our experience in the USA, but similar approaches should be applicable globally.
Direct & indirect NAb assays
There are two types of assays for NAbs: direct and indirect. The distinction is based on the drug’s mechanism of action and is tied to the choice of assay end point [4].
Direct assays are generated by drug products that exert their effect directly on a cell, stimulating responses such as receptor phosphorylation, increases in cytoplasmic ATP, cAMP, mRNA synthesis, phosphorylation of cytoplasmic proteins through specific cell-signaling pathways, cytokine production/secretion or cellular proliferation. Without NAbs, the drug product binds to a ligand-specific receptor on the cell surface and elicits a cellular response. In the presence of NAbs, that response is either decreased or abrogated.
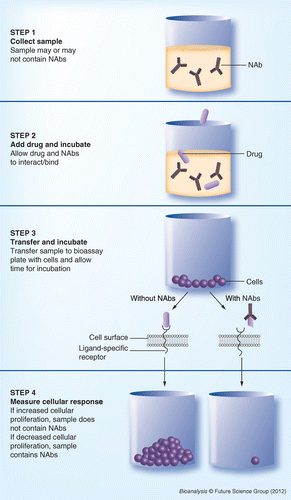
Figure 1 illustrates the main steps of a direct NAb assay using proliferation as the cellular response to the drug product. Step 1 is sample collection. At the time of collection, it is not known whether or not the sample contains NAbs. In step 2, a specific drug concentration (determined and optimized during the development phase) is added to the sample and incubated to allow any NAbs to bind to the drug product. In step 3, the sample is transferred to a bioassay plate, allowed to interact with the cells, and further incubated. In step 4, the cellular response is measured. In this example, increased cellular proliferation indicates that the drug product was able to bind to the ligand-specific receptor and that no NAbs were present in the sample. When NAbs are present they bind to the drug product, preventing it from binding to the cells and producing a response.
Indirect assays are used when the drug product (usually a monoclonal antibody or a soluble receptor) works by blocking the binding of a ligand to a specific cell-surface receptor. The desired result is a reduction or absence of a cellular response. When NAbs are present they bind to the drug product, preventing it from attaching to the cellular receptors. As a result, the ligand can bind to its promoter and trigger a specific cellular response.
The main steps for an indirect NAb assay are illustrated in Figure 2. Steps 1–3 are the same for direct and indirect assays (collect sample, add drug, transfer to bioassay plate with cells and incubate). For indirect assays, a new step (step 4) must be included to add the ligand and incubate. If the sample does not contain NAbs, the ligand-specific receptor is blocked by the drug product and the ligand itself cannot bind to the cell. If NAbs are present they bind to the drug product, leaving the ligand-specific receptor open for binding with the ligand. Step 5 measures the cellular response. In this example, the ligand triggers cellular proliferation, so a sample without NAbs exhibits decreased proliferation and a sample with NAbs results in increased cellular proliferation.
▪ Algorithms for analyzing samples
The current industry-standard tiered approach for analyzing samples as part of an immunogenicity study is depicted in Figure 3[14,15]. First, the sample is screened using a ligand-binding immunoassay, such as ELISA, to detect the presence of any drug-specific antibodies. If the sample is positive, it is then tested in a confirmatory titer test. If the sample remains positive, it is further screened using a validated NAb-specific assay. If the cellular response is normal (unaltered compared with controls), the sample does not contain NAbs and a negative result is reported.
Samples that do generate an altered cellular response (reactive samples) typically undergo further testing. Reactive samples from a direct NAb assay are verified using a confirmatory assay in which the NAbs are removed from the samples before testing. A variety of depletion techniques, such as crosslinking proteins A, G and/or L to beaded agarose or resins or chromatography, can be used. After depletion, NAb-positive samples will have diminished neutralizing capacity compared with untreated samples. In cases where NAbs need to be tested for potential cross-reactivity with the endogenous analyte (e.g., EPO), additional testing should be performed using a specificity assay with similarly diluted serum specimens that contain the endogenous analyte [16].
Since most drug products tested in an indirect NAb assay are therapeutic monoclonal antibodies that will bind to agarose beads, a different strategy must be used in the confirmatory assay. In many cases, samples are tested neat (without adding any drug or ligand). A lack of increase in cellular response (normal cell response/background) is an indication that the initial reactive sample contained NAbs. An altered cellular response indicates that the earlier result was a false positive, likely due to the presence of interfering/stimulating components in the matrix [7,14,17].
Additional consideration should be directed at factors involved in generating a nonspecific cell response that may result in a false-positive result. Cells may be responsive to multiple stimuli triggered by matrix components, such as cytokines, soluble receptors, pre-existing antibodies or endogenous proteins present as a result of a particular disease (RF factor in rheumatoid arthritis). In such cases, additional controls must be in place to ensure the specificity of the assay and lack of interference.
To measure NAbs, the current FDA guidance recommends obtaining pre-exposure samples from all patients [14]. Post-exposure sampling uses a risk-based approach that depends on a number of factors, including the frequency of dosing. Appropriate sampling intervals must be assessed on a case-by-case basis. In general, samples taken 7–14 days after drug administration will provide information on early IgM production, while samples taken 4–6 weeks following exposure are optimal for determining IgG responses.
▪ Additional information
▪ Monoclonal antibodies used for therapeutic purposes can generate NAbs. Contributing factors include antibody origin/structure (murine, chimeric, fully humanized), antibody mechanism of action and clinical variables (type of disease, age of patient, genetic factors, clinical protocol and so on) [18];
▪ These assays can be used clinically. PPD has successfully upgraded numerous cell-based NAb assays to meet Clinical Laboratory Improvement Amendments requirements for use in patient management;
▪ Preclinical studies tend to target an assay sensitivity of 500–1000 ng/ml serum, [19] while the FDA guidance suggests an assay sensitivity range of 250–500 ng/ml for most clinical trials [14].
Assay development
There are a number of parameters that are typically evaluated during the development process of NAb assays [7]. Careful planning and assay design will provide the necessary conditions for optimal and cost-efficient development (Box 1)[20].
▪ Selecting a suitable cell line
There are many factors to consider when selecting a cell line for use in a NAb assay [21]. The cell line chosen must yield a specific response to the drug product and must provide the proper sensitivity and dynamic range to measure that response. The cell line must also be able to tolerate the culture conditions and sample matrix for the drug of interest and disease state under investigation. As with all cell-based assays, the background and culture media must be carefully selected to avoid interference with the assay, and the cells must be monitored for changes induced by multiple passages. If the cell line is engineered, additional consideration must be given to specific critical areas of the process, [22,23] such as:
▪ Possible genetic drift with time and any associated loss of functionality, especially when the selective pressure is removed from the culture media;
▪ Clonal stability;
▪ Performance of the clone, ensuring consistent sensitivity and reproducibility of the assay performance;
▪ Absence of mycoplasma.
The cell-banking process involves expanding or scaling up the cell line of choice and generating a sufficient number of vials to support the validation and sample analysis processes. After scale-up, cells should be tested for mycoplasma and sterility. Use of antibiotics (such as pen-strep) in cell culture media is not advisable, because they may mask contaminations that could impact assay performance either by interacting with a component of the assay (drug product, ligand and so on) or by preventing cells from responding. As vials are usually kept in vapor-phase liquid nitrogen for long-term storage, cells should also be tested for stability in a cell-based assay to demonstrate consistency of assay performance over time.
Cell-based NAb assays that require the use of recombinant viruses necessitate purified virus preparations that must be assessed for stability and infectivity over time [24] and titered accordingly either through a TCID50 (tissue culture infective dose, or the amount of virus that infects 50% of cells in tissue culture) or a plaque assay. Finally, the multiplicity of infection needs to be determined during the development stage as this is a critical assay parameter [25].
▪ Choosing the proper cellular response (end point method)
The basis for detecting NAbs lies with the ability to observe a shift in an assay-specific cellular response. This end point must be specific and must provide the necessary sensitivity to measure changes mediated by the drug of interest. Cellular responses can be categorized as either early (receptor binding, receptor phosphorylation, detection of cAMP or ATP, signal transduction-specific protein phosphorylation) or late (cellular proliferation or apoptosis, cytokine production and reporter gene expression [fluorescence, luminescence]) events (Figure 4).
The choice of the appropriate end point method depends on the drug’s mechanism of action as well as the assay sensitivity required [7,26]. PPD has successfully used a variety of cellular responses in NAb assays: three examples are described below.
Example 1: receptor phosphorylation
In this type of cell-based assay, binding of the drug product to its receptor induces the phosphorylation of specific amino acid residues on the intracellular portion of the receptor and triggers a cascade of biological responses. To measure this response, the stimulated receptor is immuneprecipitated onto a streptavidin-coated 96-well plate from cell lysates using a biotinylated antibody against the extracellular portion of the receptor. The microtiter plate, developed by Mesoscale Discovery (Gaithersburg, MD, USA), is fitted with a series of electrodes on the bottom of each well. The captured intracellular receptor is detected using an antibody specific for the phosphorylated residues, which is further detected using a ruthenium-labeled antibody. Using the Mesoscale Discovery Sector plate reader, an electrical current is placed across the plate-associated electrodes in the presence of a buffer containing tripropylamine. The result is a series of electrically induced oxidation–reduction reactions, involving ruthenium (from the captured complex) and tripropylamine that lead to a luminescent signal. The consequent electrochemiluminescent signal is measured by photodiodes and is then quantified as a relative unit.
Example 2: detection of cell viability & cellular proliferation
Several methods for the measurement of cell viability or proliferation are available on the market. PPD has experience using CellTiter-Glo® (Promega, WI, USA), a luminescent cell assay that uses ATP to determine the number of living cells in a population. The CellTiter-Glo reagent lyses the cells, inhibits endogenous ATPases and provides luciferase/luciferin in proportion to the number of viable cells [27].
Example 3: gene-reporter assays
Many gene-reporter assays use the green fluorescent protein (in a variety of constructs [24]) and luciferase. For example, cell-based assays used to detect NAbs against interferon may be adapted from the commercially available human interferon luciferase gene-reporter iLite™ kit (PBL Interferon Source, NJ, USA). iLite cells are division-arrested, one-time use, frozen human U937 cells stably transfected with the firefly luciferase reporter gene under the control of an interferon-sensitive response element. Upon stimulation, the drug product binds to the interferon receptor on the iLite cell surface and produces luminescence by induction of the interferon-sensitive response element and, hence, the luciferase reporter gene. The luminescence intensity is proportional to the amount of drug product stimulant.
When NAbs are present, they bind to the drug product and prevent it from stimulating the cell-surface receptors and producing luminescence. Samples are scored preliminary reactive for NAbs if the luminescence readout falls below a statistically determined cut point at a single sample dilution. If the luminescence readouts are at or above the cut point, the samples are considered negative for NAbs.
▪ Selecting proper controls
One of the most important aspects of developing cell-based assays is the identification of a positive control (PC) antibody that is capable of neutralizing the biological activity of the drug product of interest. While antibodies from subjects who have tested positive for NAbs are ideal, most PC antibodies are polyclonal or monoclonal antibodies produced in animals that have been exposed to the drug product. PC antibodies are used to assess assay sensitivity, which is defined as the lowest concentration of antibodies that can be detected using the optimal drug concentration, cell density and ligand concentration (in the case of indirect assays only).
In addition to PCs, three other controls are central to the validation of NAb assays: negative control (NC), ligand control (LC) and drug control (DC). Each of these controls validates a specific component of the NAb assay to ensure that a positive result indicates the presence of NAbs and not interference from another source [7,17]. The NC uses samples from multiple drug-naive donors to test for any possible interaction between the sample matrix and the cells in culture: no drug, antibodies or ligand are present. The inclusion of donors affected by the targeted disease state is important because the disease may alter the matrix response by, for instance, increasing cytokine levels and triggering nonspecific cell stimulation [14]. In most cases, the samples are diluted before use to minimize matrix interference. The NC is also used to determine tolerance to matrix (minimum required dilution), the highest volume of sample matrix that has minimal or no impact on assay performance.
The LC, used to validate indirect assays, evaluates the potential cell stimulation caused by the ligand in the presence of the sample matrix. No drug products or drug-specific antibodies are present. Similarly, the DC is used to assess interactions between the sample matrix, cells, ligand (if appropriate) and drug in the absence of antibodies. The ratio of LC:NC (or, for direct assays, DC:NC) is a measure of the maximum stimulatory signal that can be achieved. This ratio is optimized during development and the pass/fail criterion for this ratio is established during validation. A failed LC:NC or DC:NC ratio may be indicative of problems with cells (contamination, cells kept in culture too long, wrong culture media used and so on) or the drug product (loss of potency, change in lot number and so on) and necessitates rejection of the assay.
▪ Statistical analysis of assay cut points
Most cell-based NAb assays use a statistically determined assay cut point (ACP), defined as the response level at which a sample changes from being positive to being negative (or vice versa) [7,21,28,29]. The cut point is related to the assay’s LOD, which is the smallest concentration of PC antibody that yields a positive result (Figure 5). The ACP is determined using data from at least 30 individuals who are naive to the drug of interest but, ideally, affected by the target disease [17]. The collected data are analyzed statistically to calculate a cut point with an upper negative limit of, for example, 95% (5% false positives), which assures that all positive/reactive samples will be detected [7]. Higher upper negative limits of 99% or even 99.9% may be preferred, particularly for the confirmatory cut point. The choice of the proper statistical approach should be made on a case-by-case basis, as it depends largely on the type of screening ACP used [7,17,28,29].
For the statistical analysis, the choice of parametric versus nonparametric approach depends on the distribution of data within each data panel. If, after the exclusion of outliers (either in the original scale or after appropriate transformation [log, inverse, Box-Cox transformation and so on]), the assay data for each data panel are normally distributed using a statistical test, then a parametric approach is used. If at least one assay data panel is not normally distributed and no suitable transformation can make each panel normally distributed, then a nonparametric approach should be followed [29]. While PPD commonly uses the Shapiro-Wilk test, the choice of statistical test for normality should be made in conjunction with a statistician experienced in the bioanalytical field [29,30].
A fixed screening cut point can only be used if the assay means and variances are not statistically different across data panels. If the assay means (but not the variances) are statistically different, then a floating screening cut point should be used. When the assay variances are statistically different across data panels, then a dynamic cut point is needed. In cases where the assay itself has significant inherent variability, use of a floating cut point can be considered [7,17,28,29].
▪ Optimizing assay parameters
Once the cell line and assay end point have been chosen, the assay parameters must be optimized [7,17,21]. For direct assays, both the drug concentration and the cell density require optimization. Since the sensitivity of the assay depends on the drug concentration, the lowest concentration that yields a ≥50% cellular response (ED50) within the linear portion of the curve is targeted and then further optimized to provide the best dynamic range and sensitivity. It is important to use the lowest drug concentration that provides adequate sensitivity, as higher drug concentrations may result in strong cellular responses that will not be measurably altered in the presence of low concentrations of NAbs.
Optimizing requires testing a range of drug concentrations so that the concentration-response curve plateaus at low and high concentrations, and is linear in the middle, allowing calculation of ED50 using statistical software [14]. Cell density is optimized based on signal-to-noise ratio and assay sensitivity. Indirect assays are optimized in a similar manner, first determining the lowest ligand concentration that achieves ED50 and then calculating the lowest drug concentration that causes ≥50% inhibition of the ligand. Incubation time between the drug/ligand and cells, as well as the sample minimal required dilution, must be determined to obtain the highest sensitivity while keeping background signal to a minimum.
In some cases, the presence of the drug product in matrix samples can interfere with the assay’s ability to detect NAbs. When high drug concentrations (drug tolerance) or matrix interference are expected, NAbs are often removed from the matrix with a specific binding method followed by an acid dissociation step. Using such an approach before proceeding with sample analysis allows for an accurate assessment/measurement of neutralizing activity [17,31]. PPD has successfully validated a number of cell-based NAb assays using a combination of acid dissociation and/or removal steps. However, the choice of method depends on whether the NAbs need to be removed from the matrix (high drug concentrations or interfering components in sample) or high levels of drug-bound NAbs need to be dissociated.
Other factors that can influence the assay, including cell incubation conditions, reagents or equipment and stimulation conditions, require additional development work before the assay is validated [6,17,31]. Changes in the performance of the assay (for direct or indirect assays) should also be evaluated as a function of the number of passages and/or the days in culture to avoid variability due to these factors.
▪ Assay troubleshooting
Given the nature and complexity of cell-based NAb assays, troubleshooting can be a challenging task. The most efficient approach usually involves careful study of the data generated from controls to assess potential root causes. For example, if the drug or LC fails to provide a cellular response, potential problems may include the cells themselves as well as the stability or potency of the drug product or ligand.
While troubleshooting tips could fill several volumes, many problems occur in one or a combination of these four key areas (Box 2).
▪ Validation
Method validation is the process by which an analytical procedure employed for a specific test and a specific analyte is shown to be suitable for its intended use [17,21]. Results from method validation can be used to measure the quality, reliability and consistency of analytical results. Validating NAb assays is a complex, multistep process involving several scientists in order to evaluate assay robustness. Typically, the following parameters are assessed during validation.
ACPs (screening, specificity &/or confirmatory)
The screening ACP is the screening assay value (SAV) calculated according to predetermined formulas at or above which a sample is negative and below which a sample is positive (vice versa for indirect assays), and requires further testing with a confirmatory or specificity assay. To determine the screening ACP, at least 30 individual matrix lots are analyzed three times by a minimum of two different scientists on multiple days. Statistical analysis is performed to ensure normality of the population and lack of outlier values. The specificity assay value and confirmatory assay value are determined in the same manner, with individual matrix lots analyzed three times over the course of two runs by two different scientists.
Statistical formulas may vary by assay. Examples of key formulas include, but are not limited to:
Mean response value = 
SD (σ) = 
%CV = 
SAV = 
% immunodepletion = 
PC titration (precision & sensitivity)
To determine the assay sensitivity as well as the LLOD, the PC is spiked into the pooled matrix at specific concentrations. Two curves are generated per plate and analyzed at least three times by at least two different analysts on multiple days. Precision is assessed by generating statistical analysis for between-day precision, between-curve precision and within-day precision. The lowest concentration of the PC that is below the screening ACP in all runs determines the assay sensitivity. The LLOD is defined as the lowest concentration where specific NAb antibodies can be detected 100% of the time.
Inter-assay & intra-assay precision
To assess the inter-assay precision, the PC antibody is spiked into the pooled matrix and analyzed in at least one replicate on each validation plate. To assess intra-assay precision, multiple replicates are analyzed on at least one plate. The NCs, LCs and DCs should also be monitored for precision during the validation.
LLOD (matrix recovery)
The LLOD is validated using several individual matrix samples, both untreated matrix (predose) and matrix spiked at the LLOD concentration, analyzed over the course of several runs by two scientists. The mean response, SD and %CV for each sample is calculated and reported. For each individual result, the SAV is also calculated and reported.
Confirmation of cut points
To verify that the cut points derived throughout the validation process will accurately detect NAbs in samples of the matrix of interest, all cut points should be confirmed by testing a number of individual matrix samples. Both untreated serum (predose) and serum spiked at the LLOD (postdose) are tested several times on at least two different days by at least two different analysts. Each sample is typically analyzed using both screening and confirmatory assays.
Drug interference
To assess this potential issue, the matrix is spiked with several concentrations of the drug product. The drug-spiked samples are analyzed along with multiple PC samples at concentrations above the assay LLOD to determine the amount of drug that interferes with the detection of NAbs.
Stability
The stability of NAbs is expected to be similar to the stability of any antigen-specific serum or plasma immunoglobulin, regardless of the target, and, therefore, short- and long-term stability studies for immunogenicity methods may not be necessary. However, these studies may be conducted, if needed, and mock samples (such as surrogate control spiked into matrix) can also be used.
During assay validation, matrix freeze–thaw and thawed matrix (e.g., room temperature) stability are obtained to evaluate the integrity of the controls under different short-term storage and handling conditions. To determine freeze–thaw stability of NAbs, the controls are subjected to a prespecified number of freeze–thaw cycles prior to analysis. The number of cycles depends on the needs of the tiered approach as well as future plans for the assay system. Each cycle typically consists of keeping the sample frozen for at least 24 h for the first cycle and for at least 12 h for all subsequent cycles, and then thawing the sample at room temperature. To determine room temperature stability of NAbs, the controls are allowed to stay at room temperature for at least 24 h prior to analysis.
Conclusion
As the use of therapeutic proteins increases, the risks of immunogenicity and the formation of NAbs increase as well. Cell-based assays are well-suited for detecting NAbs because they mimic the mechanism by which the NAbs and the drug interact in vivo. Each NAb assay is unique and complex, and a careful and thorough assessment and validation of multiple factors including reagents, assay controls, matrix interference and conditions will help to ensure a sensitive, specific and robust assay.
Future perspective
The technology of cell-based NAb assays has made amazing progress over the last 10 years. The next decade of scientific research will undoubtedly result in the development of increasingly more sophisticated and sensitive assays and detection systems. Key areas of anticipated improvement include:
▪ Basic science: a better and more in-depth understanding of intracellular signaling pathways should permit the development of more sophisticated engineered cell lines and detection systems;
▪ Production process: improved protein expression and purification systems and controlled post-translational modifications should result in the absence or, at least, a substantial reduction in the concentration of misfolded proteins and aggregates, as well as a decrease in the immunogenicity of therapeutic proteins;
▪ Immunogenicity prediction systems: in conjunction with in vitro cell assays and preclinical testing strategies, improved/more sophisticated prediction systems, such as computer-based evaluation/study of T-cell epitopes, should allow for the design and engineering of therapeutic molecules that are less immunogenic;
▪ Cell culture: improvement of cell culture systems, such as serum-free cultures, should help to reduce the negative or inhibitory impact on cells and improve assay sensitivity;
▪ Sensitivity: improving assay sensitivity should allow for more efficient detection of low-binding, low-affinity NAbs that may have critical clinical relevance.
Continued advances in cell-based assays will allow pharmaceutical/biotechnology companies to meet the challenges inherent to developing, testing and manufacturing novel therapeutic proteins that are generated as a result of the sequencing of the human genome.
Parameters typically evaluated during neutralizing antibody assay development.
Assay format
▪ Detailed description of the assay steps
Plate layout
▪ Usage of the outer wells
▪ Number of sample and control replicates
▪ Description/determination of the assay controls
▪ Positioning of samples and controls within a plate
Cell line
▪ Choice of the proper cell line
▪ Expected performance (sensitivity)
▪ Analytical range
▪ Cell density
▪ Drug concentration
▪ Choice of culture media and serum
▪ Tolerance/response to matrix, cell culture media and serum
▪ Determination of the optimal starvation period (if needed)
▪ Performance of the assay as a function of the number of passages/days in culture
▪ Cell banking and storage
Drug/ligand & positive control
▪ Determination of the optimal analytical range
▪ Choice of standards and QCs, if needed
▪ Determination of ‘goodness of fit’ of the curve
▪ Determination of endogenous levels in cell culture serum and matrix (if needed)
▪ Assay dynamic range
▪ Assay sensitivity
Common problems experienced with neutralizing antibody assays.
Cells/cell line stability & performance
▪ Passage number or time in culture as a function of assay performance
▪ Stability of cell surface-receptor expression (for engineered cell lines; can be assessed using flow cytometry technology)
▪ Change in cell culture media or serum (brand, lot number)
▪ Observe for any change in growth pattern or cell morphology
▪ Assess mycoplasma or other possible contamination
▪ Are banked cells properly stored? Has stability been assessed?
Reagents
▪ Stability and activity of drug product or ligand, especially if new lot is used
▪ Change in the positive control (neutralizing antibodies) or detection antibodies (species, purification process)
▪ If specialized plates are used, has there been any change in lot number? Possible lot-to-lot variability should be determined during the development process
▪ Change in lot number of detection kits
▪ Biological activity assessment of specialized reagents, such as complement
Instrumentation
▪ Change in the instrumentation parameters for the assay
▪ Maintenance or adjustment performed on the instrument
▪ Change in CO2 concentration, humidity, or temperature
▪ Introduction of new brand or model of critical equipment, such as shakers
▪ Change in pipette type (single channel vs multichannel) or tips
▪ Are the pipettes properly calibrated?
Assay format & execution
▪ Review method and ensure that steps have not been modified
▪ Change in pipetting technique
▪ Change in dilution scheme
▪ Assess assay execution by analyst
▪ Observation of assay execution by an experienced scientist
▪ Assay execution by another scientist
▪ Look for possible matrix interference
▪ Determine potential differences between healthy and diseased subjects and assess potential matrix-related interfering components
Biosimilar
Drug product that has the same therapeutic (biological) activity and is typically used at the same dose to treat the same disease as the reference product.
Neutralizing antibodies
Antibodies generated as a result of an undesired immune response following the administration of a drug product. These antibodies neutralize the biological activity of the drug product either by binding to specific sites on the drug product or by creating steric hindrance.
Enzyme-linked immunosorbent assay
Ligand-binding assay used to detect and determine the concentrations of drug products (PKs) or antibodies to drug products (immunogenicity) in a variety of matrices (both human and animal).
Therapeutic proteins (or biopharmaceuticals)
Engineered/recombinant proteins, such as monoclonal antibodies, used in the treatment of a variety of diseases/disorders, including cancers and rheumatoid arthritis.
Immunogenicity study
Experiments designed to identify the presence of drug-specific antibodies (in animal or human subjects) that could ultimately represent a risk to patient safety.
Executive summary
Background
▪ Many drug products carry the risk of generating an unwanted immune response in the patient.
▪ Factors that may increase immunogenicity include size and purity of the therapeutic protein, manufacturing process, patient health status and clinical indication, among others.
▪ This immunogenicity, which may result in the formation of antidrug antibodies, can lead to a range of clinical outcomes (from no effect to severe harm) due to altered efficacy, safety or PK of the drug.
▪ Neutralizing antibodies (NAbs), a subset of antidrug antibodies, bind to the drug product and may diminish or eliminate its biological activity. In addition to the drug product, NAbs may also cross-react with the endogenous analyte.
▪ The detection of NAbs to a particular therapeutic protein requires the development and validation of a unique cell-based assay.
Direct & indirect NAb assays
▪ Depending on the drug’s mechanism of action, NAb assays can be either direct or indirect.
▪ Direct assays are generated by drug products that exert their effect directly on a cell.
▪ Indirect assays are used when the drug works by blocking the binding of a ligand to a specific cell-surface receptor.
Assay development & validation: choosing the proper cellular response (end point method)
▪ The assay end point must be specific and must provide the necessary sensitivity to measure the cellular response mediated by the drug of interest.
Statistical analysis of assay cut points
▪ The assay cut point, which is determined statistically, is the response level at which a sample switches from being positive to being negative (or vice versa).
Optimizing assay parameters
▪ Once the cell line and assay end point have been chosen, the assay parameters (drug concentration, cell density, ligand concentration [indirect assays only]) must be optimized.
Validation
▪ Validating NAb assays is a complex, multistep process designed to demonstrate that an analytical procedure employed for a specific test and a specific analyte is suitable for its intended use.
Conclusion
▪ Each NAb assay is unique and complex, and a careful and thorough assessment and validation of multiple factors including reagents, assay controls and conditions will help to ensure a sensitive, specific and robust assay.
Acknowledgements
The authors thank R MacLean and J Harrison for critical review of the manuscript, which was prepared with the assistance of KA DeBruin.
Financial & competing interests disclosure
The authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
K DeBruin (Kodiak Consulting Group, Inc.) provided research, writing and editing services. PPD paid for her assistance.
Papers of special note have been highlighted as: ▪ of interest ▪▪ of considerable interest
References
- 1 Perini P, Facchinetti A, Bulina P et al. Interferon-beta (INF-beta) antibodies in interferon-beta1a- and interferon-beta1b-treated multiple sclerosis patients. Prevalence, kinetics, cross-reactivity and factors enhancing interferon-beta immunogenicity in vivo. Eur. Cytokine Netw.12(1),56–61 (2001).
- 2 Gilli F, van Beers M, Marnetto F, Jiskoot W, Bertolotto A, Schellenkens H. Development of a bioassay for quantification of neutralising antibodies against human interferon-beta in mouse sera. J. Immunol. Methods336(2),119–126 (2008).
- 3 Chamberlain P. Immunogenicity of therapeutic proteins. Part 1: causes and clinical manifestations of immunogenicity. Regul. Rev.5(5),2–9 (2002).
- 4 Chamberlain P. Immunogenicity of therapeutic proteins. Part 2: measurement, prediction, minimisation and regulatory consequences of immunogenicity. Regul. Rev.5(6),4–10 (2002).
- 5 Porter S. Human immune response to recombinant human proteins. J. Pharm. Sci.90(1),1–11 (2001).▪ Review of immune responses observed during development of recombinant human proteins.
- 6 Lee JW, Kelley M, King LE et al. Bioanalytical approaches to quantify ‘total’and ‘free’ therapeutic antibodies and their targets: technical challenges and PK/PD applications over the course of drug development. AAPS J.139(1),99–110 (2011).
- 7 Gupta S, Indelicato SR, Jethwa V et al. Recommendations for the design, optimization, and qualification of cell-based assays used for the detection of neutralizing antibody responses elicited to biological therapeutics. J. Immunol. Methods321(1–2),1–18 (2007).▪▪ Recommendations on developing cell-based assays for neutralizing antibodies.
- 8 Rosenberg AS. Effects of protein aggregates: an immunologic perspective. AAPS J.8(3),E501–E507 (2006).
- 9 Cromwell ME, Hilario E, Jacobson F. Protein aggregation and bioprocessing. AAPS J.8(3),E572–E579 (2006).
- 10 Ponce R, Abad L, Amaravadi L et al. Immunogenicity of biologically-derived therapeutics: assessment and interpretation of nonclinical safety studies. Regul. Toxicol. Pharmacol.54(2),164–182 (2009).▪ Description of factors contributing to the generation of drug-specific antibodies.
- 11 Browne SK, Brubelo PD, Chetchotisakd P et al. Adult-onset immunodeficiency in Thailand and Taiwan. N. Engl. J. Med.367(8),725–734 (2012).
- 12 Casadevall N, Nataf J, Viron B et al. Pure red-cell aplasia and antierythropoietin antibodies in patients treated with recombinant erythropoietin. N. Engl. J. Med.346(7),469–475 (2002).
- 13 Sanchez S. Barger T, Zhou L et al. Strategy to confirm the presence of anti-erythropoietin neutralizing antibodies in human serum. J. Pharm. Biomed. Anal.55(5),1265–1274 (2011).
- 14 US FDA. Guidance for Industry: Assay Development for Immunogenicity Testing of Therapeutic Proteins. FDA, Silver Spring, MD, USA (2009).▪▪ US Government guidance for development of immunogenicity assays.
- 15 Committee for Medicinal Products for Human Use. Guideline on Immunogenicity Assessment of Biotechnology-Derived Therapeutic Proteins. European Medicines Agency, London, UK (2006).▪▪ European Government guidance for development of immunogenicity assays.
- 16 MacLean RC, Leu K, Jolicoeur P, Tacey R, Green J, Schatz P. Validation of a cell-based assay for the quantification of Hematide™-specific neutralizing antibodies in clinical serum specimens. Presented at: 2009 AAPS National Biotechnology Conference. Seattle, WA, USA, 21–24 June 2009.
- 17 Gupta S, Devanarayan V, Finco D et al. Recommendations for the validation of cell-based assays used for the detection of neutralizing antibody immune responses elicited against biological therapeutics. J. Pharm. Biomed. Anal.55(5),878–888 (2011).▪▪ Recommendations on validating cell-based assays for neutralizing antibodies.
- 18 Committee for Medicinal Products for Human Use. Guideline on Immunogenicity Assessment of Monoclonal Antibodies Intended for In Vivo Clinical Use. European Medicines Agency, London, UK (2010).
- 19 Lofgren JA, Wala I, Koren E, Swanson SJ, Jing S. Detection of neutralizing anti-therapeutic protein antibodies in serum or plasma samples containing high level of therapeutic protein. J. Immunol. Methods308(1–2),101–108 (2006).
- 20 Chen XC, Zhou L, Gupta S, Civoli F. Implementation of design of experiments (DOE) in the development and validation of a cell-based bioassay for the detection of anti-drug neutralizing antibodies in human serum. J. Immunol. Methods376(1–2),32–45 (2012).
- 21 Kelley M. Considerations while setting up cell-based assays. In: Validation of Cell-Based Assays in the GLP Setting: A Practical Guide. Prabhakar U, Kelley M (Eds). John Wiley & Sons, Ltd., West Sussex, UK, 1–10 (2008).▪▪ Practical information on developing cell-based assays.
- 22 Grimm S. The art and design of genetic screens: mammalian culture cells. Nat. Rev. Genet.5(3),179–189 (2004).
- 23 Wurm FM. Production of recombinant protein therapeutics in cultivated mammalian cells. Nat. Biotechnol.22(11),1393–1398 (2012).
- 24 MacLean R, Jolicoeur P, Tacey R. The development and validation of a cell-based assay for the detection of neutralizing antibodies against MVA-X in human serum. Presented at: 2012 AAPS National Biotechnology Conference. San Diego, CA, USA, 21–23 May 2012.
- 25 Merten O-W, Al-Rubeai M. Viral vectors for gene therapy: methods and protocols. In: Methods in Molecular Biology. Humana Press, NY, USA, 737 (2011).
- 26 Deyes R, Arduengo M, Allard STM. Choosing the right assay to monitor your signal transduction pathway. Cell Notes21,13–16 (2008).
- 27 MacLean RC, Jolicoeur P, Tacey R et al. Development and validation of a cell-based assay for the detection of Valortim® in New Zealand white (NZW) rabbit serum samples. Presented at: 2012 AAPS National Biotechnology Conference. San Diego, CA, USA, 21–23 May 2012.
- 28 Mire-Sluis AR, Barrett YC, Devanarayan V et al. Recommendations for the design and optimization of immunoassays used in the detection of host antibodies against biotechnology products. J. Immunol. Methods289(1–2),1–16 (2004).▪ Practical information on cut point calculation.
- 29 Shankar G, Devanarayan V, Amaravadi L et al. Recommendations for the validation of immunoassays used for detection of host antibodies against biotechnology products. J. Pharm. Biomed. Anal.48(5),1267–1281 (2008).▪ Practical information on cut point calculation.
- 30 Razali NM, Wah YB. Power comparisons of Shapiro-Wilk, Kolmogorov-Smirnov, Lilliefors and Anderson-Darling tests. J. Stat. Modeling Analyt.2(1),21–33 (2011).
- 31 Smith HW, Butterfield A, Sun D. Detection of antibodies against therapeutic proteins in the presence of residual therapeutic protein using a solid-phase extraction with acid dissociation (SPEAD) sample treatment prior to ELISA. Regul. Toxicol. Pharmacol.49(3),230–237 (2007).


