







Discovery of indolyl-containing peptides as novel antibacterial agents targeting tryptophanyl-tRNA synthetase
Abstract
Background: There is an urgent need for antibiotics with novel structures and unexploited targets to counteract bacterial resistance. Methodology & results: Novel tryptophanyl-tRNA synthetase inhibitors were discovered based on virtual screening, surface plasmon resonance binding, enzymatic activity assay and antibacterial activity evaluation. Of the 29 peptide derivatives tested for antibacterial activity, some inhibited the growth of both Staphylococcus aureus and Staphylococcus epidermidis. A13 and A15 exhibited antibacterial activity against methicillin-resistant S. aureus NRS384 at an 8 μg/ml minimum inhibitory concentration. A13 snugly docked into the active site, explaining its improved inhibitory activity. Conclusion: Our results provide us with new structural clues to develop more potent tryptophanyl-tRNA synthetase inhibitors and lay a solid foundation for future drug design efforts.
Long-term and excessive use of antibiotics for the treatment of bacterial infections in both humans and animals have led to the emergence of drug-resistant bacteria [1,2]. Antibiotic resistance is becoming increasingly serious and causes significant damage to human health [3]. An estimated 9.3 million people died as a result of microbial infection in 2015, which surpassed the number of deaths caused by cancer [4]. Thus, exploring antimicrobials with novel mechanisms or chemical entities is strongly recommended to curtail this crisis [5,6]. The aaRS family is one such group that has recently garnered more attention for the development of small molecule inhibitors [7,8].
aaRSs are essential enzymes that play a central role in the correct translation of genetic information into proteins [9]. Each aaRS catalyze a specific amino acid to attach to the cognate tRNA molecule in the presence of ATP, yielding an aminoacyl-tRNA for further use in protein synthesis. Therefore, aaRS have proven vital in obtaining the necessary translational accuracy [10]. Inhibition of any enzyme will lead to protein synthesis inhibition and cell growth arrest. Furthermore, these enzymes display significant divergence between prokaryotic and eukaryotic cells in the topology and functions of the binding domain. This indicates that aaRSs are considered a promising target for antimicrobial agent development. This has been further exemplified by the clinical use of mupirocin (isoleucyl-tRNA synthetase inhibitor), which is marketed by GSK as Bactroban [11,12]. In addition, tavaborole (formerly AN2690), as an editing site inhibitor of leucyl-tRNA synthetase, was approved by the US FDA in July 2014 for the treatment of onychomycosis [13].
However, a limited number of TrpRS inhibitors have been reported previously. Chuangxinmycin and indolmycin are natural products isolated from Actinoplanes tsinanensis [14] and the African strain of Streptomyces albus [15], respectively (Figure 1). Chuangxinmycin was initially reported for its moderate activity against a range of Gram-positive bacteria, such as Staphylococcus aureus (minimum inhibitory concentration [MIC]: 4-8 μg/ml). Indolmycin bears some resemblance to chuangxinmycin, although it was reported to have better antibacterial activity against staphylococci (MIC: 0.5–2 μg/ml) [16]; however, further development was discontinued since it appeared that indolmycin was not sufficiently active against the majority of commonly occurring pathogenic bacteria such as Streptococci, Enterococci and Enterobacteriaceae [17]. Furthermore, certain strains that are resistant toward both compounds have been isolated [18].

Substrate mimic-based modification is another way to find new aaRS inhibitors. In general, the aminoacyl-adenylate (aa-AMP) intermediate has a two or three order of magnitude higher affinity for the enzyme compared with the substrate amino acid or ATP [8,19]. However, many of the aa-AMP intermediates lack antibacterial activity. Furthermore, most of the aa-AMP modifications face issues including chemical stability, binding affinity, uptake problems and selectivity [19]. Recently, Arthur van Aerschot’s group reported a sulfamate bridge to 3-deazaadenosine aa-AMP analogs; while chemical stability improved, the compounds unfortunately lacked antibacterial activity in the whole cell [20].
Therefore, the discovery of more small-molecule TrpRS inhibitors is still urgently needed to supplement the available structural arsenal and antibacterial design based on the target.
Nowadays, virtual screening plays an indispensable role in the discovery of novel small-molecule inhibitor to the target protein. The primary advantage of the method is its high screening speed. However, this method suffers from a high false positive rate. Furtherly, surface plasmon resonance (SPR) binding and enzymatic activity assay can validate the direct interaction with the target in vitro. Antibacterial activity assay will finally be used to evaluate the compounds in the whole cell. Clearly, these methods complement each other will increase the speed and accuracy of screening results; Hence, combining these approaches will be a valid strategy to find novel TrpRS inhibitors. We expect the new findings may provide us with structural clues to develop more potent antibacterial agents.
Materials & methods
Virtual screening & activity verification
Virtual screening
In order to discover more TrpRS inhibitors, we virtually screened a combined library based on Life Chemicals library ( www.lifechemicals.com) and our in-house small molecule library containing over 25,000 compounds. The active site was defined as all the amino acid residues confined within a 9 Å radius sphere centered about the Escherichia coli TrpRS crystal complex ligand (Protein Data Base [PDB] ID: 5V0I). After LibDockScore screening, the 2000 top-ranked compounds obtained using the libdockscore function were re-docked using the CDOCKER mode. A total of 341 compounds could not dock into the active site, so the remaining 1659 compounds were selected for visual inspection. Then, 76 compounds with reasonable poses were selected to undergo the next examination for chemical diversity in order to limit redundancy in our initial testing. Finally, 38 compounds were selected to examine their direct interaction with TrpRS and undergo a biological evaluation.
Activity verification
A total of 38 compounds were screened by SPR, including 35 compounds purchased from the Life Chemicals library and three synthesized compounds, A0, A1 and A2 (
The 35 purchased compounds were diluted to 100 μM in phosphate-buffered saline P20 Plus (PBS-P+) buffer and then injected across the CM5 chip. Chuangxinmycin and ofloxacin were used as the positive and negative controls, respectively. Compounds F5038-0132, F5831-1969, F6414-3612, F2704-0451 with response binding signals higher than a third of the positive control were re-tested in a dose-dependent assay. Compounds A0, A1 and A2 underwent a direct binding test by SPR.
Of the tested compounds, only F5831-1969, F6414-3612, F2704-0451, A0 (
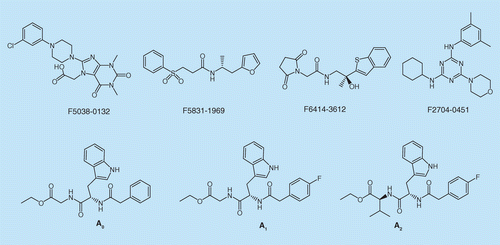
Encouraged by their direct interaction with the protein, the inhibitory activity of the six compounds were further evaluated on Gram-positive (S. aureus NRS384) and Gram-negative (E. coli ATCC25922) bacterial strains. Fortunately, Compound A2 possessed a novel tripeptide structure could inhibit the growth of S. aureus NRS384 at 32 μg/ml. Hence, A2 was identified as a hit compound for further optimization.
Derivative structure design
Compound A2 can be divided into three parts as shown in Figure 3. There was an isopropyl in part I. In order to inspect the size and hydrophobic/hydrophilic activity of this substituent, compounds A0–A12 were designed and synthesized. Additionally, compounds B1–B3 with different ester groups were synthesized to evaluate the differences in activity.

The scaffold was divided into part I (pink), part II (blue) and part III (red).
Given that both the substrate tryptophan and reported TrpRS inhibitors have an indolyl group in their structure, we reserved this structure in most of the compounds except A21, in which structural/property analogous phenolic hydroxyl group was instead. In natural or synthesized compounds, steric configuration always plays a vital role in biological activity. Four compounds C2, C3, C6 and C8 possessing a D-tryptophanyl group were designed to test whether the configuration affected antibacterial activity.
In addition, there was a fluorine atom in the para-position of benzene in part III, so compounds A13–A20 with different electron-withdrawing or electron-donating groups were synthesized to investigate the structure–activity relationship (SAR) on part III.
Chemistry
29 compounds were designed and synthesized using the routes outlined in Figure 4. In order to investigate the influence of part I and the configuration of tryptophanate, condensation of 1 with 2a (ethyl L-tryptophanate hydrochloride) and 2b (ethyl D-tryptophanate hydrochloride) in the presence of HATU (O-(7-aza-1H-benzotriazol-1-yl)-N,N,N’,N’-tetramethyluronium hexafluorophosphate) and DIEA (N,N-diisopropylethylamine) followed by hydrolysis with LiOH in water and THF (tetrahydrofuran) yield 4a and 4b, which were condensated with the corresponding amino acid ester hydrochloride, to yield target compounds A1–12 and C2,3,6,8 (Figure 4 protocol 1).


In protocol 1, compounds were synthesised from part III to II then I. In protocol 2 synthesis was from I to II then finally to part III.
DIEA: N,N-diisopropylethylamine; DMF: Dimethylformamide; HATU: O-(7-aza-1H-benzotriazol-1-yl)-N,N,N’,N’-tetramethyluronium hexafluorophosphate; rt: Room temperature; THF: Tetrahydrofuran.
Compound A6 was hydrolyzed to yield compound 6. Target compounds B1,2,3 were obtained by esterification of compound 6 with the corresponding alcohol in the presence of SOCl2.
In order to investigate the influence of part III, condensation of 5f with 8 gave intermediate 9. The protective group was then removed in the presence of diethylamine. The desired products A13–20 were obtained by condensation with the corresponding aromatic carboxylic acid. Compounds A0 and A21 were obtained with the same protocol using ethyl glycinate hydrochloride (5a) and Fmoc-L-tryptophan or fmoc-L-tyrosine as starting materials, respectively (Figure 4 Protocol 2).
Materials & general information
All reagents and anhydrous solvents were obtained from commercial sources and used without further purification. Indolmycin was purchased from Alfa chemistry (NY, USA). Melting points (mp) were determined in open capillaries using a Mettler Toledo MP90 melting point system (Mettler Toledo, Shanghai, China). 1H-nuclear magnetic resonance (NMR; 400 MHz) and 13C-NMR (101 MHz) spectra were recorded on a Bruker Avance (Bruker, Karlsruhe, Germany) in DMSO-d6 using tetramethylsilane (TMS) as an internal standard. Electrospray ionization high-resolution mass spectra (ESI-HRMS) were recorded using a Thermo LTQ ORBITRAP XL mass spectrometer (Thermo Fisher Scientific, MA, USA). Intermediate mass spectrometry data were reported using liquid chromatography–mass spectrometry (LC–MS; Shimadzu, Shanghai, China) with a gradient of 0.1% formic acid in water (MP-A) and 0.1% formic acid in acetonitrile (MP-B) from 10% MP-B to 90% MP-B over 5 min, then maintained for 3 min with a flow rate of 0.4 ml/min. Flash column chromatography was performed on a Combiflash Rf 200, 0.038 mm particle size. Thin-layer chromatography (TLC) was performed on silica gel plates (ART5554 60F254; Merck, Darmstadt, Germany).
Compounds A1–A12, B1–B3, C2, C3, C6 and C8 were prepared as shown in protocol 1; A0 and A13–A21 were prepared as shown in protocol 2 (Figure 4). The synthesis of the target compounds (A0, A1, A2, A3, A6, A8, A9, A10, C2, B1, A13, A14, A15, and A21) was shown as follows. The synthesis of the rest target compounds and all the intermediates was present in the Supporting data.
General procedure for the preparation of compounds A1–A12, C2, C3, C6, & C8
To a solution of compound 4a/4b (150 mg, 0.44 mmol) in anhydrous dimethylformamide (DMF, 7 ml), 1.05 equivalents of amino acid ester hydrochloride, 1.1 equivalents of HATU and 2 equivalents of DIEA was successively added. The reaction mixture was stirred for 4 h at room temperature. 50 ml of water was added and extracted by ethyl acetate (3 × 30 ml). The combined organic layers were washed with water and brine (40 ml), then dried over Na2SO4, filtered and concentrated. The residue was purified by flash chromatography using MeOH:DCM (0–3:97) as eluent to provide the A1–12, C2, C3, C6, and C8 as a white solid.
Ethyl(2-(4-fluorophenyl)acetyl)-L-tryptophylglycinate (A1)
Following the general procedure, employing 4a and ethyl glycinate hydrochloride 5a as starting material, yielded compound A1 as a white solid in 68% yield, mp: 166.8–168.1°C. 1H-NMR (400 MHz, DMSO-d6): δ 10.81 (s, 1H), 8.53 (t, J = 5.6 Hz, 1H), 8.27 (d, J = 8.4 Hz, 1H), 7.60 (d, J = 8.0 Hz, 1H), 7.33 (d, J = 8.0 Hz, 1H), 7.11 (d, J = 1.6 Hz, 1H), 7.08–7.04 (m, 3H), 7.02–6.95 (m, 3H), 4.59 (td, J = 8.8, 4.4 Hz, 1H), 4.09 (q, J = 7.2 Hz, 2H), 3.90–3.79 (m, 2H), 3.44–3.40 (m, 2H), 3.15 (dd, J = 14.8, 4.4 Hz, 1H), 2.92 (dd, J = 14.8, 9.2 Hz, 1H), 1.19 (t, J = 7.2 Hz, 3H). 13C-NMR (101 MHz, DMSO-d6) δ: 172.18, 169.74, 169.71, 162.83 (d, J = 242.4 Hz), 136.05, 132.40 (d, J = 3.0 Hz), 130.74, 130.66, 127.27, 123.70, 120.81, 118.42, 118.19, 114.77, 114.56, 111.24, 109.98, 60.41, 53.15, 41.08, 40.78, 27.99, 14.07; ESI-HRMS (m/z): calcd for C23H25O4N3F [M+H]+: 426.18236, found: 426.18198.
Ethyl(2-(4-fluorophenyl)acetyl)-L-tryptophyl-L-valinate (A2)
Following the general procedure, employing 4a and ethyl L-valinate hydrochloride 5b as starting material, yielded compound A2 as a white solid in 67% yield, mp: 63.1–64.9°C. 1H-NMR (400 MHz, DMSO-d6): δ 10.81 (s, 1H), 8.24 (d, J = 8.0 Hz, 2H), 7.60 (d, J = 7.6 Hz, 1H), 7.32 (d, J = 8.0 Hz, 1H), 7.10–7.06 (m, 4H), 7.04–6.95 (m, 3H), 4.69 (td, J = 8.8, 4.8 Hz, 1H), 4.18–4.16 (m, 1H), 4.13–4.05 (m, 2H), 3.42–3.34 (m, 2H), 3.11 (dd, J = 14.4, 4.8 Hz, 1H), 2.92 (dd, J = 14.4, 9.2 Hz, 1H), 2.08–1.99 (m, 1H), 1.18 (t, J = 7.2 Hz, 3H), 0.87 (t, J = 6.0 Hz, 6H). 13C-NMR (101 MHz, DMSO-d6): δ 172.00, 171.25, 169.74, 160.85 (d, J = 242.4 Hz), 136.01, 132.42 (d, J = 3.0 Hz), 130.70, 130.63, 127.35, 123.63, 120.79, 118.52, 118.12, 114.79, 114.58, 111.19, 110.94, 60.32, 57.49, 52.93, 41.08, 29.89, 27.78, 18.88, 18.23, 14.10. ESI-HRMS (m/z): calcd for C26H31O4N3F [M+H]+: 468.22931, found: 468.22873.
Ethyl(2-(4-fluorophenyl)acetyl)-L-tryptophyl-L-methioninate (A3)
Following the general procedure, employing 4a and ethyl L-methioniate hydrochloride 5c as starting material, yielded compound A3 as a yellow solid in 61% yield, mp: 146.1–147.5°C. 1H-NMR (400 MHz, DMSO-d6): δ 10.81 (s, 1H), 8.47 (d, J = 7.6 Hz, 1H), 8.25 (d, J = 8.4 Hz, 1H), 7.61 (d, J = 7.6 Hz, 1H), 7.32 (d, J = 8.0 Hz, 1H), 7.11 (d, J = 2.0 Hz, 1H), 7.09–7.04 (m, 3H), 7.02–6.95 (m, 3H), 4.59 (td, J = 8.8, 4.8 Hz, 1H), 4.41–4.35 (m, 1H), 4.13–4.04 (m, 2H), 3.42–3.37 (m, 2H), 3.13 (dd, J = 14.8, 4.8 Hz, 1H), 2.92 (dd, J = 14.8, 9.6 Hz, 1H), 2.47–2.39 (m, 2H), 2.02 (s, 3H), 2.00–1.84 (m, 2H), 1.17 (t, J = 7.2 Hz, 3H). 13C-NMR (101 MHz, DMSO-d6): δ 171.99, 171.57, 169.72, 160.8 (d, J = 245.4 Hz), 136.03, 132.43 (d, J = 2.0 Hz), 130.69, 130.61, 127.28, 123.67, 120.82, 118.46, 118.15, 114.78, 114.57, 111.22, 109.92, 60.58, 53.05, 51.06, 41.05, 30.49, 29.49, 27.82, 14.58, 14.04; ESI-HRMS (m/z): calcd for C26H31O4N3FS [M+H]+: 500.20138, found: 500.20085.
Methyl(S)-2-((S)-2-(2-(4-fluorophenyl)acetamido)-3-(1H-indol-3-yl)propanemido)pentanoate (A6)
Following the general procedure, employing 4a and methyl (S)-2-aminopentanoate hydrochloride 5f as starting material, yielded compound A6 as a white solid in 85% yield, mp: 166.4–167.7°C; 1H-NMR (400 MHz, DMSO-d6): δ 10.81 (s, 1H), 8.39 (d, J = 7.6 Hz, 1H), 8.23 (d, J = 8.4 Hz, 1H), 7.61 (d, J = 7.6 Hz, 1H), 7.32 (d, J = 8.0 Hz, 1H), 7.10 (d, J = 2.0 Hz, 1H), 7.09–7.04 (m, 3H), 7.02-6.95 (m, 3H), 4.62 (td, J = 8.8, 4.8 Hz, 1H), 4.29–4.23 (m, 1H), 3.61 (s, 3H), 3.41–3.37 (m, 2H), 3.11 (dd, J = 14.8, 4.8 Hz, 1H), 2.91 (dd, J = 14.8, 9.2 Hz, 1H), 1.69–1.54 (m, 2H), 1.33–1.23 (m, 2H), 0.85 (t, J = 7.2 Hz, 3H); 13C-NMR (101 MHz, DMSO-d6): δ 172.52, 171.85, 169.69, 160.84 (d, J = 242.4 Hz), 136.02, 132.45 (d, J = 3.0 Hz), 130.70, 130.62, 127.32, 123.69, 120.80, 118.52, 118.13, 114.78, 114.57, 111.19, 109.91, 52.98, 51.79, 51.70, 41.07, 32.87, 27.84, 18.58, 13.46; ESI-HRMS m/z: calcd for C25H29O4N3F [M+H]+: 454.21366, found: 454.21384.
Methyl(S)-2-((S)-2-(2-(4-fluorophenyl)acetamido)-3-(1H-indol-3-yl)propanamido)-3,3-dimethylbutanoate (A8)
Following the general procedure, employing 4a and methyl (S)-2-amino-3,3-dimethylbutanoate hydrochloride 5h as starting material, yielded compound A8 as a white solid in 90% yield, mp: 59.8–61.2°C 1 H-NMR (400 MHz, DMSO-d6): δ 10.81 (s, 1H), 8.27 (d, J = 8.0 Hz, 1H), 8.06 (d, J = 8.8 Hz, 1H), 7.57 (d, J = 7.6 Hz, 1H), 7.32 (d, J = 8.0 Hz, 1H), 7.11–7.06 (m, 4H), 7.03–6.94 (m, 3H), 4.72 (td, J = 8.8, 5.2 Hz, 1H), 4.18 (d, J = 8.8 Hz, 1H), 3.61 (s, 3H), 3.42–3.38 (m, 2H), 3.08 (dd, J = 14.8, 4.8 Hz, 1H), 2.96–2.92 (m, 1H), 0.89 (s, 9H); 13C-NMR (101 MHz, DMSO-d6): δ 171.81, 171.15, 169.88, 160.87 (d, J = 242.4 Hz), 135.99, 132.39 (d, J = 3.0 Hz), 130.71, 130.63, 127.34, 123.60, 120.79, 118.50, 118.13, 114.81, 114.60, 111.18, 109.90, 60.23, 52.94, 51.45, 41.07, 33.77, 27.47, 26.40; ESI-HRMS m/z: calcd for C26H31O4N3F [M+H]+: 468.22931, found: 468.22833.
Methyl (2-(4-fluorophenyl)acetyl)-L-tryptophyl-L-serinate (A9)
Following the general procedure, employing 4a and methyl O-(tert-butyl)-L-serinate hydrochloride 5i as starting material, yielded compound A9–0. Then removal of the tertiary butyl protective group yielded compound A9. To a solution of compound A9–0 (250 mg, 0.50 mmol) in dichloromethane (DCM, 9 ml), 70 equivalents of trifluoroacetic acid was added. The reaction mixture was stirred for 2 h at room temperature, monitored by TLC (DCM:MeOH = 10:1), and then concentrated. The residue was dissolved in 30 ml ethyl acetate, 20 ml water. Then the pH was adjusted to 6–7 by saturated sodium bicarbonate solution and it was extracted by ethyl acetate (2 × 30 ml). The combined organic layers were washed with water and brine (40 ml), dried over Na2SO4, filtered and concentrated. The residue was purified by flash chromatography using MeOH:DCM (0–8:92) as eluent to provide the A9 as a a white solid in 69% yield mp: 205.2–207.1°C 1H-NMR (400 MHz, DMSO-d6): δ 10.80 (s, 1H), 8.41 (d, J = 7.6 Hz, 1H), 8.22 (d, J = 8.4 Hz, 1H), 7.62 (d, J = 8.0 Hz, 1H), 7.32 (d, J = 8.0 Hz, 1H), 7.10 (d, J = 1.6 Hz, 1H), 7.07–7.03 (m, 3H), 7.01–6.94 (m, 3H), 5.05 (t, J = 5.6 Hz, 1H), 4.66 (td, J = 8.8, 5.2 Hz, 1H), 4.39–4.35 (m, 1H), 3.74–3.69 (m, 2H), 3.63 (s, 3H), 3.41–3.34 (m, 2H), 3.14 (dd, J = 14.8, 4.4 Hz, 1H), 2.91 (dd, J = 14.8, 9.2 Hz, 1H), 1.70–1.55 (m, 2H), 1.33–1.22 (m, 2H), 0.85 (t, J = 7.2 Hz, 3H); 13C-NMR (101 MHz, DMSO-d6): δ 171.90, 170.93, 169.67, 160.82 (d, J = 242.4 Hz), 136.02, 132.40 (d, J = 2.0 Hz), 130.71, 130.63, 127.35, 123.77, 120.77, 118.55, 118.13, 114.78, 114.57, 111.18, 109.93, 61.07, 54.76, 53.04, 51.87, 41.06, 27.98; ESI-HRMS m/z: calcd for C23H25O5N3F [M+H]+: 442.17728, found: 442.17710.
Methyl (2-(4-fluorophenyl)acetyl)-L-tryptophyl-L-lysinate A10
Following the general procedure, employing 4a and N-Boc-L-Lysine methyl ester hydrochloride 5j as starting material, yielded compound A10–0 then removed the Boc protective group yielded compound A10. To a solution of compound A10–0 (300 mg, 0.52 mmol) in DCM (10 ml), 50 equivalents of trifluoroacetic acid and 2 equivalents of triethylsilane was added. The reaction mixture was stirred for 4 h at -3°C, monitored by TLC (DCM:MeOH = 10:1) and then concentrated at room temperature. The residue was dissolved in 30 ml ethyl acetate, then 20 ml saturated sodium bicarbonate solution was added, extracted by ethyl acetate (2 × 30 ml), dried over Na2SO4, filtered and concentrated. The residue was purified by flash chromatography using MeOH:DCM (0–12:88) as eluent to provide the A10 as a a colorless oil in 62% yield. 1H-NMR (400 MHz, DMSO-d6): δ 10.85 (s, 1H), 8.45 (d, J = 7.6 Hz, 1H), 8.29–8.25 (m, 1H), 7.61 (d, J = 7.6 Hz, 1H), 7.53 (brs, 2H), 7.33 (d, J = 8.0 Hz, 1H), 7.12–6.95 (m, 7H), 4.61–4.56 (m, 1H), 4.29–4.24 (m, 1H), 3.62 (s, 3H), 3.41–3.37 (m, 2H), 3.13 (dd, J = 14.4, 4.4 Hz, 1H), 2.92 (dd, J = 14.4, 9.6 Hz, 1H), 2.73 (t, J = 7.2 Hz, 2H), 1.76–1.59 (m, 2H), 1.51-1.44 (m, 2H), 1.35–1.30 (m, 2H); 13C-NMR (101 MHz, DMSO-d6): δ 172.37, 171.95, 169.80, 160.85 (d, J = 242.4 Hz), 136.05, 132.43 (d, J = 3.0 Hz), 130.71, 130.63, 127.29, 123.75, 120.81, 118.50, 118.15, 114.81, 114.60, 111.24, 109.85, 53.16, 51.87, 51.78, 41.04, 38.69, 30.24, 27.82, 26.80, 22.28; ESI-HRMS m/z: calcd for C26H32O4N4F [M+H]+: 483.24021, found: 483.23981.
Ethyl(2-(4-fluorophenyl)acetyl)-D-tryptophyl-L-valinate C2
Following the general procedure, employing 4b and ethyl L-valinate hydrochloride 5b as starting material, yielded compound C2 as a white solid in 78% yield, mp: 58.2–60.1°C; 1H-NMR (400 MHz, DMSO-d6): δ 10.80 (s, 1H), 8.33 (d, J = 8.4 Hz, 1H), 8.27 (d, J = 8.4 Hz, 1H), 7.62 (d, J = 8.0 Hz, 1H), 7.31 (d, J = 8.0 Hz, 1H), 7.12–6.94 (m, 7H), 4.77–4.71 (m, 1H), 4.14–4.03 (m, 3H), 3.43–3.34 (m, 2H), 3.08 (dd, J = 14.4, 5.2 Hz, 1H), 2.92 (dd, J = 14.4, 8.8 Hz, 1H), 2.00–1.92 (m, 1H), 1.17 (t, J = 7.2 Hz, 3H), 0.77 (t, J = 6.8 Hz, 6H); 13C-NMR (101 MHz, DMSO-d6): δ 171.91, 171.44, 169.62, 160.85 (d, J = 242.4 Hz), 136.01, 132.47 (d, J = 3.0 Hz), 130.70, 130.63, 127.32, 123.72, 120.78, 118.78, 118.10, 114.78, 114.57, 111.15, 109.83, 60.34, 57.42, 53.13, 41.08, 29.95, 28.59, 18.84, 18.10, 14.08; ESI-HRMS m/z: calcd for C26H31O4N3F [M+H]+: 468.22931, found: 468.22925.
General procedure for the preparation of compounds B1–B3
The corresponding alcohol was cooled to 0°C, 12 equivalents of SOCl2 was added and keeping the solution at 0°C for 20 min. Then compound 6 (250 mg, 0.57 mmol) was added. The reaction mixture was stirred for 5 h at room temperature and monitored by TLC (DCM:MeOH = 10:1), then it was quenched by 30 ml saturated sodium bicarbonate solution and extracted by ethyl acetate (3 × 30 ml). The combined organic layers were washed with water and brine, dried over Na2SO4, filtered and concentrated. The residue was purified by flash chromatography using MeOH:DCM (0–2:98) as eluent to provide the B1–B3 as a white solid.
Ethyl(S)-2-((S)-2-(2-(4-fluorophenyl)acetamido)-3-(1H-indol-3-yl)propanamido)pentanoate B1
Following the general procedure, employing ethyl alcohol 7a as starting material, yielded compound B1 as a white solid in 79% yield, mp: 166.8–167.9°C; 1H-NMR (400 MHz, DMSO-d6): δ 10.81 (s, 1H), 8.39 (d, J = 7.6 Hz, 1H), 8.23 (d, J = 8.4 Hz, 1H), 7.62 (d, J = 7.6 Hz, 1H), 7.33 (d, J = 8.0 Hz, 1H), 7.11 (d, J = 1.6 Hz, 1H), 7.09–7.04 (m, 3H), 7.02–6.95 (m, 3H), 4.63 (td, J = 8.8, 4.4 Hz, 1H), 4.26–4.20 (m, 1H), 4.12–4.04 (m, 2H), 3.41–3.37 (m, 2H), 3.12 (dd, J = 14.8, 4.8 Hz, 1H), 2.91 (dd, J = 14.8, 9.6 Hz, 1H), 1.71–1.55 (m, 2H), 1.35–1.24 (m, 2H), 1.17 (t, J = 7.2 Hz, 3H), 0.86 (t, J = 7.2 Hz, 3H); 13C-NMR (101 MHz, DMSO-d6): δ 172.01, 171.90, 169.69, 160.84 (d, J = 242.4 Hz), 136.04, 132.45 (d, J = 3.0 Hz), 130.70, 130.62, 127.32, 123.69, 120.80, 118.52, 118.14, 114.77, 114.56, 111.20, 109.96, 60.39, 52.95, 51.85, 41.08, 32.87, 27.90, 18.60, 14.07, 13.50; ESI-HRMS m/z: calcd for C26H31O4N3F [M+H]+: 468.22931, found: 468.22995.
General procedure for the preparation of compounds A0, A13-A21
To a solution of (0.6 mmol) compound 10, 13 or 16 in anhydrous DMF (8 ml), 1.05 equivalents of aromatic carboxylic acid, 1.1 equivalents of HATU and 2 equivalents of DIEA was successively added, then stirred for 4 h at room temperature. The reaction was monitored by TLC (DCM:MeOH = 10:1), quenched by H2O (50 ml) and extracted by ethyl acetate (3 × 30 ml). The combined organic layers were washed with water and brine (40 ml), dried over Na2SO4, filtered and concentrated. The residue was purified by flash chromatography using MeOH:DCM (0-3:97) as eluent to provide the A0, A13–A21 as a white solid.
Methyl(S)-2-((S)-2-(2-(3-fluorophenyl)acetamido)-3-(1H-indol-3-yl)propanamido)pentanoate A13
Following the general procedure, employing compound 10 and 2-(3-fluoro-phenyl)acetic acid (11m) as starting material, yielded compound A13 as a white solid in 82% yield, mp: 168.1–169.6°C; 1H-NMR (400 MHz, DMSO-d6): δ 10.81 (s, 1H), 8.40 (d, J = 7.6 Hz, 1H), 8.29 (d, J = 8.0 Hz, 1H), 7.61 (d, J = 8.0 Hz, 1H), 7.32 (d, J = 8.0 Hz, 1H), 7.26–7.21 (m, 1H), 7.11 (d, J = 1.6 Hz, 1H), 7.06 (t, J = 7.6 Hz, 1H), 7.02–6.89 (m, 4H), 4.62 (td, J = 8.8, 5.2 Hz, 1H), 4.29–4.24 (m, 1H), 3.61 (s, 3H), 3.46–3.39 (m, 2H), 3.12 (dd, J = 14.4, 4.8 Hz, 1H), 2.91 (dd, J = 14.4, 9.2 Hz, 1H), 1.67–1.56 (m, 2H), 1.33–1.23 (m, 2H), 0.85 (t, J = 7.2 Hz, 3H); 13C-NMR (101 MHz, DMSO-d6): δ 172.51, 171.80, 169.28, 161.92 (d, J = 244.4 Hz), 139.07 (d, J = 8.1 Hz), 136.02, 129.81 (d, J = 9.1 Hz), 127.32, 125.06 (d, J = 3.0 Hz), 123.62, 120.79, 118.50, 118.13, 115.67 (d, J = 21.2 Hz), 112.99 (d, J = 21.2 Hz), 111.21, 109.89, 53.05, 51.78, 51.68, 41.56, 32.87, 27.84, 18.56, 13.44; ESI-HRMS m/z: calcd for C25H29O4N3F [M+H]+: 454.21366, found: 454.21249.
Methyl(S)-2-((S)-2-(2-(2-fluorophenyl)acetamido)-3-(1H-indol-3-yl)propanamido)pentanoate A14
Following the general procedure, employing compound 10 and 2-(2-fluoro-phenyl)acetic acid (11n) as starting material, yielded compound A14 as a white solid in 76% yield, mp: 157.9–159.8°C; 1H-NMR (400 MHz, DMSO-d6): δ 10.83 (s, 1H), 8.38 (d, J = 7.6 Hz, 1H), 8.27 (d, J = 8.0 Hz, 1H), 7.62 (d, J = 8.0 Hz, 1H), 7.32 (d, J = 8.0 Hz, 1H), 7.26–7.21 (m, 1H), 7.12 (d, J = 2.0 Hz, 1H), 7.12–7.00 (m, 4H), 6.97 (t, J = 7.6 Hz, 1H), 4.63 (td, J = 8.8, 5.2 Hz, 1H), 4.30–4.24 (m, 1H), 3.61 (s, 3H), 3.51–3.40 (m, 2H), 3.12 (dd, J = 14.4, 4.8 Hz, 1H), 2.92 (dd, J = 14.4, 8.8 Hz, 1H), 1.69–1.56 (m, 2H), 1.34–1.24 (m, 2H), 0.86 (t, J = 7.2 Hz, 3H); 13C-NMR (100 MHz, DMSO-d6): δ 172.52, 171.78, 168.79, 160.45 (d, J = 244.4 Hz), 136.02, 131.47 (d, J = 5.1 Hz), 128.39 (d, J = 8.1 Hz), 127.35, 124.00 (d, J = 3.0 Hz), 123.65, 123.19 (d, J = 16.2 Hz), 120.80, 118.49, 118.14, 114.85 (d, J = 21.2 Hz), 110.20, 109.90, 53.08, 51.78, 51.71, 34.84, 32.89, 27.83, 18.58, 13.47; ESI-HRMS m/z: calcd for C25H29O4N3F [M+H]+: 454.21366, found: 454.21432.
Methyl(S)-2-((S)-3-(1H-indol-3-yl)-2-(2-phenylacetamido)propanamido)pentanoate A15
Following the general procedure, employing compound 10 and 2-phenylacetic acid (11o) as starting material, yielded compound A15 as a white solid in 82% yield, mp: 156.1–156.8°C; 1H-NMR (400 MHz, DMSO-d6): δ 10.81 (s, 1H), 8.38 (d, J = 7.2 Hz, 1H), 8.22 (d, J = 8.0 Hz, 1H), 7.61 (d, J = 7.6 Hz, 1H), 7.32 (d, J = 8.0 Hz, 1H), 7.21–7.16 (m, 2H), 7.10 (d, J = 1.6 Hz, 1H), 7.08–7.04 (m, 2H), 6.97 (t, J = 7.6 Hz, 1H), 4.62 (td, J = 8.8, 5.2 Hz, 1H), 4.29–4.23 (m, 1H), 3.61 (s, 3H), 3.43–3.36 (m, 2H), 3.12 (dd, J = 14.8, 4.8 Hz, 1H), 2.91 (dd, J = 14.8, 9.2 Hz, 1H), 1.70–1.55 (m, 2H), 1.33–1.22 (m, 2H), 0.85 (t, J = 7.2 Hz, 3H); 13C-NMR (101 MHz, DMSO-d6): δ 172.52, 171.86, 169.79, 136.30, 136.02, 128.91, 128.02, 127.34, 126.13, 123.67, 120.78, 118.51, 118.12, 111.18, 109.91, 52.99, 51.78, 51.69, 42.03, 32.88, 27.83, 18.57, 13.47; ESI-HRMS m/z: calcd for C25H30O4N3 [M+H]+: 436.22308, found: 436.22418.
Ethyl (2-phenylacetyl)-L-tryptophylglycinate A0
Following the general procedure, employing 13 and 2-phenylacetic acid (11o) as starting material, yielded compound A0 as a white solid in 78% yield, mp: 155.1–156.9°C; 1H-NMR (400 MHz, DMSO-d6): δ 10.82 (s, 1H), 8.54 (t, J = 5.6 Hz, 1H), 8.28 (d, J = 8.0 Hz, 1H), 7.61 (d, J = 7.6 Hz, 1H), 7.32 (d, J = 8.0 Hz, 1H), 7.21–7.12 (m, 4H), 7.06–7.04 (m, 3H), 6.97 (t, J = 7.6 Hz, 1H), 4.59 (td, J = 8.8, 4.8 Hz, 1H), 4.09 (q, J = 7.2 Hz, 2H) 3.90–3.78 (m, 2H), 3.45–3.35 (m, 2H), 3.15 (dd, J = 14.8, 4.4 Hz, 1H), 2.93 (dd, J = 14.8, 9.2 Hz, 1H), 1.19 (t, J = 7.2 Hz, 3H); 13C-NMR (101 MHz, DMSO-d6): δ 172.23, 169.86, 169.73, 136.26, 136.06, 129.98, 128.03, 127.29, 126.14, 123.72, 120.82, 118.44, 118.19,111.25, 110.00, 60.43, 53.17, 42.05, 40.80, 28.01, 14.09; ESI-HRMS m/z: calcd for C23H26O4N3 [M+H]+:408.19178, found: 408.19189.
Ethyl(2-(4-fluorophenyl)acetyl)-L-tyrosylglycinate A21
Following the general procedure, employing 16 and 2-(4-fluorophenyl)acetic acid (1) as starting material, yielded compound A21 as a white solid in 69% yield, mp: 156.4–158.1°C; 1H-NMR (400 MHz, DMSO-d6): δ 9.17 (s, 1H), 8.48 (t, J = 5.6 Hz, 1H), 8.27 (d, J = 8.8 Hz, 1H), 7.08–6.99 (m, 6H), 6.61 (d, J = 8.4 Hz, 1H), 4.47 (td, J = 10.0, 4.0 Hz, 1H), 4.09 (q, J = 7.2 Hz, 2H), 3.89–3.78 (m, 2H), 3.43–3.31 (m, 2H), 2.92 (dd, J = 13.6, 4.0 Hz, 1H), 2.62 (dd, J = 13.6, 10.0 Hz, 1H), 1.19 (t, J = 7.2 Hz, 3H); 13C-NMR (101 MHz, DMSO-d6): δ 171.97, 169.71, 169.68, 160.85 (d, J = 243.4 Hz), 155.77, 132.45, 130.74, 130.66, 130.09, 127.86, 114.82, 114.74, 114.53, 60.41, 54.00, 41.13, 40.72, 36.98, 14.06; ESI-HRMS m/z: calcd for C21H24O5N2F [M+H]+: 403.16638, found: 403.16690.
SPR-based binding assays
SPR-binding assays were performed on a Biacore T200 instrument (GE Healthcare, MA, USA). TrpRS protein (20 μg/ml) was injected across ligand flow channels for covalent immobilization on a CM5 chip. Then, the chip was equilibrated with PBS-P+ buffer (20 mM phosphate buffer, 2.7 mM KCl, 137 mM NaCl, 0.05% P20 surfactant, and 5% (v/v) DMSO).
For affinity measurement, compound stock were diluted at concentrations ranging from 200 to 3.125 μM in the running buffer (PBS-P+ buffer, 5% DMSO, pH 7.4) and injected at a flow rate of 30 μl/min for a 60 s association phase, followed by up to a 300 s dissociation phase. The state model of T200 evaluation software was used to analyze the resulting data to calculate the KD of the compounds.
Enzymatic activity assay of TrpRS
Enzymatic activity was measured using a Kinase-Glo Plus Luminescent Kinase Assay (Promega, WI, USA) by quantifying the amount of ATP remaining in solution after the reaction as reported previously [21]. According to the protocol, reactions were carried out in solid white opaque, flat-bottomed 96-well plates. A 50 µl total volume of the reaction mixture containing the compound, Bacillus stearothermophilus TrpRS, MgCl2 and dithiothreitol (DTT) dissolved with Tris (pH 8.0) buffer was used to make twofold serial dilutions. Then, a 25 μl mixture containing L-tryptophan and ATP was added. The final 50 μl reaction mixture contained 1 μM B. stearothermophilus TrpRS, 16 μM ATP, 40 mM Tris (pH 8.0), 8 mM MgCl2, 0.8 mM DTT, 1 mM L-tryptophan and 0–25 μM compound. After incubation at 37°C for 90 min, an equal volume of Kinase-Glo reagent was added to each well, mixed, and kept at 25°C for 60 min before the final recording of luminescence on a SYNERGY H1 microplate reader (Biotech, VT, USA). The rate of adenylation inhibition by the compound was calculated using the following formula:
The half maximal inhibitory concentration (IC50) value of each compound was determined by GraphPad Prism 5 software.
Antimicrobial activity
The antibacterial activities of the synthesized compounds were tested against strains from the American Type Culture Collection (ATCC) and clinical isolates from Chinese hospitals. Vancomycin and meropenem were used as positive controls for the Gram-positive and Gram-negative bacterial strains, respectively, using Mueller–Hinton broth medium (Oxoid, Hampshire, UK). Amphotericin B was used as a positive control for fungi using RPMI1640 medium (Procell Life Science and Technology Co., Wuhan, China). A stock solution of the synthesized compound (1280 μg/ml) in DMSO was prepared. MICs of the target compounds were determined using the broth dilution assay at various concentrations (128.0, 64.0, 32.0, 16.0, 8.0, 4.0, 2.0, 1.0, 0.5, 0.25, 0.125 and 0.06 μg/ml) based on the standard liquid dilution of CLSI M07-A9. Microorganism suspensions were prepared to contain approximately 105 CFU/ml and applied to 96-well plates. Culture plates were incubated at 37°C for 24 h. In the case of fungi, plates were incubated at 35°C for 24 h. MICs were defined as the lowest concentrations that prevented visible microorganism growth.
Docking study
In order to inspect and compare the binding mode of compound A13 with indolmycin to the TrpRS, the crystal structure of TrpRS (PDB code 5DK4) was downloaded and modified by adding hydrogen atoms and removing H2O molecules except reserving H2O 525, 589, 591 and Mg2+ which playing an important role in stabilizing the state of the enzyme using Discovery Studio 3.0 software. The active site was defined as all the amino acid residues confined within a 9 Å radius sphere centered about indolmycin. After the protein assigned CHARMm, compound A13 was docked using the CDOCKER module. Top eight poses were exported for visual inspection and further analysis.
Results & discussion
Antibacterial activity
The evaluation results of the antimicrobial activities of all synthetic compounds against representative Gram-positive bacteria (S. aureus ATCC 29213 and S. aureus NRS384) and Gram-negative bacteria (E. coli ATCC25922) are presented in Table 1.
  | |||||||
|---|---|---|---|---|---|---|---|
| Compound | R1 | R2 | R3 | R4 | MIC (μg/ml) | ||
| A† | B‡ | C§ | |||||
| A0 | -C2H5 | -H | -H |  | >128 | >128 | >128 |
| A1 | -C2H5 | -H | 4-F |  | >128 | >128 | >128 |
| A2 | -C2H5 | -CH(CH3)2 | 4-F |  | 32 | >128 | >128 |
| A3 | -C2H5 |  | 4-F |  | 32 | >128 | >128 |
| A4 | -C2H5 | -CH3 | 4-F |  | >128 | >128 | >128 |
| A5 | -CH3 | -CH2CH3 | 4-F |  | 128 | 128 | >128 |
| A6 | -CH3 |  | 4-F |  | 16 | 32 | >128 |
| A7 | -CH3 |  | 4-F |  | >128 | >128 | >128 |
| A8 | -CH3 |  | 4-F |  | 32 | >128 | >128 |
| A9 | -CH3 |  | 4-F |  | >128 | >128 | >128 |
| A10 | -CH3 |  | 4-F |  | >128 | >128 | >128 |
| A11 | -C2H5 |  | 4-F |  | >128 | >128 | >128 |
| A12 | -C2H5 |  | 4-F |  | >128 | >128 | >128 |
| A13 | -CH3 |  | 3-F |  | 8 | 16 | >128 |
| A14 | -CH3 |  | 2-F |  | 16 | 64 | >128 |
| A15 | -CH3 |  | 4-H |  | 8 | 32 | >128 |
| A16 | -CH3 |  | 4-Cl |  | >128 | >128 | >128 |
| A17 | -CH3 |  | 4-Br |  | >128 | >128 | >128 |
| A18 | -CH3 |  | 4-OH |  | 64 | 128 | >128 |
| A19 | -CH3 |  | 4-NO2 |  | >128 | >128 | >128 |
| A20 | -CH3 |  | 4-OCH3 |  | >128 | >128 | >128 |
| A21 | -C2H5 | -H | 4-F |  | >128 | >128 | >128 |
| B1 | -C2H5 |  | 4-F |  | 32 | >128 | >128 |
| B2 |  |  | 4-F |  | >128 | >128 | >128 |
| B3 |  |  | 4-F |  | >128 | >128 | >128 |
| C2 | -C2H5 | -CH(CH3)2 | 4-F |  | >128 | >128 | >128 |
| C3 | -C2H5 |  | 4-F |  | >128 | >128 | >128 |
| C6 | -CH3 |  | 4-F |  | >128 | >128 | >128 |
| C8 | -CH3 |  | 4-F |  | >128 | >128 | >128 |
| Indolmycin | 1 | 1 | 128 | ||||
| Vancomycin | 1 | 1 | – | ||||
| Meropenem | – | – | 0.03125 | ||||
| Compound | MIC (μg/ml) | |||||||
|---|---|---|---|---|---|---|---|---|
| D† | E‡ | F§ | G¶ | H# | I†† | J‡‡ | K§§ | |
| A2 | 128 | 64 | >128 | 128 | 128 | >128 | 128 | 128 |
| A3 | >128 | >128 | >128 | >128 | >128 | >128 | >128 | >128 |
| A5 | 128 | 128 | >128 | 128 | 128 | 128 | 128 | 128 |
| A6 | 128 | 32 | 64 | >128 | 128 | 128 | 128 | 128 |
| A8 | >128 | 128 | >128 | 128 | 128 | 128 | 128 | 128 |
| A13 | 128 | 128 | 64 | 128 | 128 | 128 | 128 | 128 |
| A14 | 128 | 64 | 128 | 128 | 128 | 128 | 128 | 128 |
| A15 | 64 | 64 | 64 | 128 | 128 | 128 | 128 | 128 |
| A18 | 128 | 64 | >128 | 128 | 128 | 128 | 128 | 128 |
| B1 | >128 | >128 | >128 | >128 | >128 | >128 | >128 | >128 |
| Vancomycin | 2 | 1 | 1 | – | – | – | – | – |
| Meropenem | 4 | 2 | 4 | 0.5 | <0.03 | – | – | – |
| Amphotericin B | – | – | – | – | – | 0.5 | 0.25 | 1 |
| Indolmycin | 0.5 | 0.25 | 0.5 | >128 | 128 | 128 | 128 | 128 |
Additionally, antibacterial activity of the compounds against other strains including three Gram-positive bacteria (methicillin-resistant Staphylococcus epidermidis 17–1, methicillin-sensitive S. epidermidis 17–1, and methicillin-resistant S. aureus ATCC 33591) using vancomycin as the reference control, two Gram-negative bacteria (Shigella 17–1 and Proteus mirabillis 17–1) using Meropenem as the positive control and three fungi (Candida albicans ATCC 90029, Candida glabrata KTCC 0001, and Candida krusei ATCC 6258) using amphotericin B as the positive control are presented in Table 2.
In general, many of the evaluated compounds showed activity against Gram-positive bacteria, but all completely lost antibacterial activity against E. coli ATCC25922.
Of the tested compounds, A6 exhibited antibacterial activity toward S. aureus NRS384 and S. aureus ATCC 29213, with MIC values of 16 and 32 μg/ml, respectively. Replacement of the fluorine atom at the meta-position (A13) or fluorine removal (A15) increased compound potency and resulted in an MIC value of 8 μg/ml toward S. aureus NRS384, which was less potent than indolmycin (MIC: 1 μg/ml) and could inhibit the growth of S. aureus ATCC 29213 at 16–32 μg/ml. The corresponding hydroxyl at the para-position (A18) significantly reduced potency, with an MIC value of 64 μg/ml for S. aureus NRS384 and a loss of activity against S. aureus ATCC 29213. However, replacement of the fluorine atom at the para-position with chlorine (A16) or bromine (A17) led to a loss of antibacterial activity. Replacement of fluorine with methoxyl (A20) or nitro (A19) groups were also detrimental to biochemical activity.
We turned our attention to exploring the SAR profile of the R2 substituent in hopes of finding a permissive region. In comparison with A2, substitution with H (A1) or methyl (A4) led to activity loss. Substitution with 3-sulphur substituted butyl (A3) showed activity toward S. aureus NRS384 (MIC value of 32 μg/ml). Replacement of the isopropyl with tert-butyl resulted in an equally potent compound (A8).
Of these compounds, A6 showed the best activity. Lengthening (A7) or shortening (A5) the hydrophobic chain resulted in activity loss. Furthermore, exchange of the hydrophobic chain containing 3–4 carbons to rigid aryl (A11 and A12) or hydrophilic (A9 and A10) groups resulted in activity loss. This suggests that the substituent at this position interacts with a small hydrophobic area of the enzyme.
We then focused our efforts on investigating the influence of the ester group. Exchange of the methyl ester to ethyl ester resulted in potency attenuation, while replacement of the ethyl ester with n-propyl ester (B2) or isopropyl ester (B3) led to activity loss, suggesting a larger ester group was not tolerated.
Interestingly, the configuration of tryptophanate is crucial to activity. Compared witrh L-tryptophanate, the D-diastereoisomers (C2. C3. C6. and C8) completely lost activity.
Compound A6 showed activity toward clinical isolates S. epidermidis 17–1 (MIC value of 32 μg/ml) that was better than those of compounds A2, A14, A15 and A18 whose MIC values were 64 μg/ml. In addition, compounds A6, A13 and A15 showed better activity toward methicillin-resistant S. aureus ATCC 33591, consistent with the results against S. aureus NRS384. However, in Gram-negative bacteria and fungi such as E. coli, Shigella and Candida, antibacterial activity was lost. We speculated that the outer membrane exists outside the cell walls may prevent the compounds from reaching intracellular targets.
SPR-based binding assay
Given the different antibacterial activity between compounds, SPR-based binding assay was performed to detect the differences in binding affinity between compounds (Figure 5). First, we turned our attention to the substituent of R2. In contrast to A1, substitution with 3-sulphur substituted butyl (A3) and isopropyl (A2) improved binding affinity tenfold. Compounds A2 and A3 inhibited S. aureus NRS384 growth at 32 μg/ml. The binding between compound A6 with n-propyl in position R2 and TrpRS with a KD value of 4.6 μM was approximately 100-fold more potent than that of the corresponding unsubstituted compound A1. In addition, the MIC value of A6 was 16 μg/ml.
The binding affinity of A13, Indolmycin and TrpRS was less potent than A6 (
In compound A11, substitution with rigid benzyl led to a near complete loss of binding ability (
We then turned our attention to part II. Compared with A1, replacement of the indolyl group with the phenolic hydroxyl group in compound A21 led to an almost complete loss in binding ability, indicating the importance of the indolyl group in protein binding (
Inhibitory activities against TrpRS
In order to investigate compound enzymatic activity, B. stearothermophilus TrpRS was overexpressed and purified. Compounds with antibacterial activity were measured, and the results were shown in Table 3.The inhibitory activity of the represent compounds was shown in Figure 6.
| Compound | A2 | A3 | A6 | A8 | A13 | A14 | A15 | A18 | B1 | Indolmycin |
|---|---|---|---|---|---|---|---|---|---|---|
| IC50 (μM) | 7.19 | 17.00 | 13.04 | 8.55 | 11.49 | >50 | 17.84 | >50 | 14.38 | 5.67 |
Most of the active compounds inhibited enzymatic activity in a dose-dependent manner. The IC50 values of the compounds were 7–18 μM, which was slightly weaker than the TrpRS inhibitor indolmycin (IC50 5.67 μM). The IC50 value of A18 was above 50 μM, this was consistent with it having the lowest antibacterial activity of the tested compounds. However, there was a discrepancy between enzyme inhibition activity and antibacterial activity for compound A14; the MIC value of A14 was 16 μg/ml and the IC50 value of A14 was above 50 μM. Ortho F could affect the enzymatic activity of the compound. We speculate that there may exist another mechanism affecting antibacterial activity that warrants further investigation. Overall, the enzymatic activity data demonstrated that these compounds acted as antimicrobial agents by inhibiting TrpRS activity.
Docking studies
Molecular docking studies were performed to explain the compounds at the molecular level, and compare them to the docking mode of the known TrpRS inhibitor indolmycin. The most active compound A13 was docked into the active site of B. stearothermophilus-derived TrpRS-based on the binding model of the indolmycin–TrpRS complex structure (5dk4.pdb) [22].

The KD value of A1, A2, A3 and A6 was 2.787 × 10-4 M, 3.326 × 10-5 M, 3.078 × 10-5 M and 4.653 × 10-6 M, respectively.

The half maximal inhibitory concentration value of A2, A6, A13 and indolmycin was 7.19, 13.04, 11.49 and 5.67 μM, respectively; without inhibitors, the luminescence value of A2, A6, A13 and indolmycin was 139089, 112437, 136291 and 109216 RLU, respectively.
The binding model of compound A13 and TrpRS is depicted in Figure 7A & B. Several hydrogen-bonding interactions together with some hydrophobic interactions tightly anchoring A13 to the active site might explain its inhibitory activity.

(A) Binding mode of compound A13 with TrpRS from Bacillus stearothermophilus. For clarity, only interacting residues were labeled. (B) 2D interaction of A13 and TrpRS. Key bonds are indicated by dashed lines between the atoms involved. The colors of key bonds and residues are shown according to interaction mode. (C) Alignment of A13 (magenta) and indolmycin (green) at the active site. TrpRS is shown in white. (D) Close-up view of the hydrophobic pocket. A13 is depicted in magenta, the surrounding key residues are shown in green and labeled. These images were produced using Discovery Studio 3.0 software.
In brief, the alignments for A13 and indolmycin in the active site are shown in Figure 7C. The planar indole moiety of compound A13 and indolmycin were arranged in nearly the same position, which can be identified by the same amino acid residue Asp132. Another hydrogen-bonding interaction was formed between the N of the imidazole ring and His43 which can enhance this interaction.
The carbonyl group of the tryptophanyl moiety also provided a considerable contribution to the binding affinity by forming three important interactions with the protein. Carbonyl O formed a typical hydrogen bond with the sidechain NH2 group of Gln9. It also formed a hydrogen bond and metal-interaction with H2O and Mg2+, respectively, which play an important role in the conformational stability of the enzyme [23,24]. These findings confirm that this carbonyl group is crucial for binding.
In part I, there are two important hydrogen bonds in the compound; one is between the NH and the carbonyl O of Gly7, while the other is between the carbonyl O atom of the norvalyl and the Asn18 sidechain NH2 group.
Furthermore, hydrophobic interactions are evident in the binding of A13 with TrpRS. The indole-ring of the molecule matches the π–π interaction with His43 and the π–alkyl interaction with Val40 and Mse129. As shown in Figure 7D, the methyl and n-butyl groups in part I drop completely into a hydrophobic pocket built by Val143, Gly144, Val141, Ala22, Ile8 and Phe26, n-butyl groups involved in the hydrophobic interactions with Ile8, Phe26 and Ala22, which indicates a detriment of a hydrophilic substituent in these positions. Therefore, the substitution of a methylene hydroxyl and n-butylamino group resulted in a considerable decrease in potency. This docking model also revealed that increasing the hydrophobic groups in these positions may also cause a steric hindrance to the Ala22 and Phe26 residue, leading to reduced binding affinity. For a definite conclusion, modification at these position with a hydrophobic group owning suitable steric hindrance would improve antibacterial activity.
Conclusion
In this study, we identified A2 as a potential TrpRS inhibitor-based on virtual screening. Enzymatic activity assay confirmed the inhibitory activity with an IC50 value of 7.19 μM. SPR assay demonstrated the direct binding between A2 and TrpRS. The indolyl group played an important role in protein binding. Based on the scaffold, a series of derivatives were designed and synthesized to evaluate their antibacterial activity. Compounds A13 and A15 exhibited antibacterial activity against methicillin-resistant S. aureus NRS384 with MIC values of 8 μg/ml, which was weaker than that of Indolmycin. A2, A6, A14, A15 and A18 inhibited the growth of methicillin-sensitive S. epidermidis 17–1 at 32–64 μg/ml. Furthermore, A6, A13 and A15 also showed antibacterial activity toward methicillin-resistant S. aureus ATCC 33591 at 64 μg/ml. Unfortunately, the designed compound could not inhibit Gram-negative bacteria or fungi. Compared with Gram-positive bacteria, there is an outer membrane existing outside the cell walls of Gram-negative bacteria, such as E. coli; the outer membrane is composed of lipopolysaccharides, phospholipids, lipoproteins et al. In fungi, the structure of the cell wall is much denser and firmer than that of Gram-positives. We hypothesized that the outer membrane of negative bacteria and the cell wall of fungi constitute a barrier for the entry of molecules, which may prevent the compounds from targeting the TrpRS in the cytoplasm and thus had no bacteriostatic effect.
The inhibitory activity of active compounds A2, A3, A6, A8, A13, A15 and B1 against TrpRS were validated, with IC50 values of 7–18 μM, The IC50 value of A18 was higher than 50 μM, consistent with its lower antibacterial activity. Compound A14 inhibited the growth of S. aureus NRS384 at 16 μg/ml. In contrast, the enzymatic activity was relatively lower, indicating that another mechanism may exist that affects antibacterial activity and requires further investigation.
Compound A13 was docked into the active site of B. stearothermophilus-derived TrpRS which was reported by Williams et al [22]. It occupied the substrate pocket. The indole ring of A13 and indolmycin were arranged in nearly the same position. As proposed by Williams, the hydrogen bond network of hexavalent complexes involved by Mg2+, H2O and indolmycin played an important role in stabilization protein conformation, compound A13 could also form interaction with H2O and Mg2+ which can also contribute to the stability of the protein. Molecular docking study revealed a hydrophobic R2 group with suitable steric hindrance is essential to improving activity. Taken together, these novel tripeptides provide us with new structural clues to develop more potent TrpRS inhibitors and lay a solid foundation for discovering novel antibacterial agents.
Future perspective
Antibiotic-resistant pathogen infection poses an ever-increasing threat to public health. The discovery of novel antibacterial candidates has recently entered a bottleneck period. However, as the details of resistance mechanisms are disclosed, more potential targets will be exposed. In addition to the classic tools of synthetic chemistry, microbiology and chemical biology, computer-aided drug design will be vastly more targeted and intelligent. The multidisciplinary approaches of the research programs will make the discovery of novel antibacterial candidates much more efficient.
The novel structure of bacterial TrpRS inhibitor was discovered by virtual screening and activity verification.
29 tripeptide derivatives were designed and synthesized.
The antibacterial activities of all compounds were evaluated against Escherichia coli and Staphylococcus aureus.
The active compounds were further evaluated against Staphylococcus epidermidis, Shigella, Proteus mirabilis and three fungi.
Compound A13 and A15 exhibited antibacterial activity against methicillin-resistant S. aureus NRS384 with MIC values of 8 μg/ml.
A2, A6, A14, A15 and A18 could inhibit the growth of methicillin-sensitive S. epidermidis 17–1 at 32–64 μg/ml.
An surface plasmon resonance-based binding assay demonstrated the direct binding between compounds and the target.
The enzymatic activity data demonstrated the inhibitory activity of the compounds toward the bacterial TrpRS.
Molecular docking of A13 indicated that the inhibitor snug fit of the inhibitor in the active site may well explain its improved antibacterial activity.
These novel tripeptides provide us with new structural clues to develop more potent TrpRS inhibitors and novel antibacterial agent.
Supplementary data
To view the supplementary data that accompany this paper please visit the journal website at: www.future-science.com/doi/suppl/10.4155/fmc-2020-0016
Acknowledgments
The authors acknowledge Dr Zhaoyong Yang (Institute of Medicinal Biotechnology, Chinese Academy of Medical Sciences and Peking Union Medical College) for providing Bacillus stearothermophilus TrpRS and Sichuan Primed Bio-Tech Group Co., Ltd for aid with compounds antibacterial activity testing.
Financial & competing interests disclosure
This work was supported by grants from the CAMS Innovation Fund for Medical Sciences (no. 2017-I2M-1-012) and grant from the National Natural Science Foundation of China (no. 81773585). The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
Open access
This work is licensed under the Attribution-NonCommercial-NoDerivatives 4.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
Papers of special note have been highlighted as: • of interest; •• of considerable interest
References
- 1. . General principles of antibiotic resistance in bacteria. Drug Discov. Today Technol. 11, 33–39 (2014).
- 2. . Molecular mechanisms of antibiotic resistance. Chem. Commun. 47(14), 4055–4061 (2011).
- 3. Bad bugs, no drugs: no ESKAPE! an update from the Infectious Diseases Society of America. Clin. Infect. Dis. 48(1), 1–12 (2009).
- 4. . Counting the dead and what they died from: an assessment of the global status of cause of death data. Bull. World Health Organ. 83(3), 171–177 (2005).
- 5. . Antibacterial drug discovery in the resistance era. Nature 529(7586), 336–343 (2016). • Very important report regarding the current situation of multidrug resistance and antibacterial drug discovery.
- 6. . Antimicrobial resistance mechanisms and potential synthetic treatments. Future Sci. OA 4(4), FSO290 (2018).
- 7. . Prospects for aminoacyl-tRNA synthetase inhibitors as new antimicrobial agents. Antimicrob. Agents Chemother. 49(12), 4821–4833 (2005).
- 8. . Aminoacyl-tRNA synthetases: structure, function, and drug discovery. Int. J. Biol. Macromol. 111, 400–414 (2018). • Very important report regarding aaRS as the target of antibacterial agent.
- 9. . Aminoacyl-tRNA synthetases: general features and recognition of transfer RNAs. Annu. Rev. Biochem. 48, 601–648 (1979).
- 10. . Aminoacyl-tRNA synthesis. Annu. Rev. Biochem. 69, 617–650 (2000).
- 11. . Evolutionary limitation and opportunities for developing tRNA synthetase inhibitors with 5-binding-mode classification. Life 5(4), 1703–1725 (2015).
- 12. . Aminoacyl tRNA synthetases as targets for new anti-infectives. FASEB J. 12(15), 1599–1609 (1998).
- 13. An antifungal agent inhibits an aminoacyl-tRNA synthetase by trapping tRNA in the editing site. Science 316(5832), 1759–1761 (2007).
- 14. The antimicrobial natural product chuangxinmycin and some synthetic analogues are potent and selective inhibitors of bacterial tryptophanyl tRNA synthetase. Bioorg. Med. Chem. Lett. 12(21), 3171–3174 (2002).
- 15. . PA 155A, B, and X antibiotics produced by a strain of Streptomyces albus. Antibiot. Chemother. 10, 316–320 (1960).
- 16. . Anti-staphylococcal activity of indolmycin, a potential topical agent for control of staphylococcal infections. J. Antimicrob. Chemother. 54(2), 549–552 (2004).
- 17. . Aminoacyl-tRNA synthetase inhibitors as potential antibiotics. Eur. J. Med. Chem. 46(11), 5227–5236 (2011). •• Very important review regarding aaRS inhibitors as potential antibiotics.
- 18. . Indolmycin resistance of Streptomyces coelicolor A3(2) by induced expression of one of its two tryptophanyl-tRNA synthetases. J. Biol. Chem. 277(26), 23882–23887 (2002).
- 19. . 5’-(N-aminoacyl)-sulfonamido-5’-deoxyadenosine: attempts for a stable alternative for aminoacyl-sulfamoyl adenosines as aaRS inhibitors. Eur. J. Med. Chem. 93, 227–236 (2015). • Very important report regarding aminoacyl-adenylate mimics as aaRS inhibitors.
- 20. Family-wide analysis of aminoacyl-sulfamoyl-3-deazaadenosine analogues as inhibitors of aminoacyl-tRNA synthetases. Eur. J. Med. Chem. 148, 384–396 (2018).
- 21. Potent and selective inhibitors of Staphyllococcus epidermidis tryptophanyl-tRNA synthetase. J. Antimicrob. Chemother. 60(3), 502–509 (2007). •• Reported the method regarding enzymatic activity assay of TrpRS.
- 22. . Selective inhibition of bacterial tryptophanyl-tRNA synthetases by indolmycin is mechanism-based. J. Biol. Chem. 291(1), 255–265 (2016). •• Very important report regarding the crystal structure of indolmycin and bacterial TrpRS.
- 23. . A master switch couples Mg2+-assisted catalysis to domain motion in B. stearothermophilus tryptophanyl-tRNA synthetase. Structure 20(1), 128–138 (2012). • Important report regarding the protein conformational changes of bacterial TrpRS in the process of binding small molecular inhibitors.
- 24. . Independent saturation of three TrpRS subsites generates a partially assembled state similar to those observed in molecular simulations. Proc. Natl Acad. Sci. USA 106(6), 1790–1795 (2009).


