







Peptides for specific intracellular delivery and targeting of nanoparticles: implications for developing nanoparticle-mediated drug delivery
Abstract
The use of peptides to mediate the delivery and uptake of nanoparticle (NP) materials by mammalian cells has grown significantly over the past 10 years. This area of research has important implications for the development of new therapeutic materials and for the emerging field of NP-mediated drug delivery. In this review, we highlight recent advances in the delivery of various NPs by some of the more commonly employed cellular delivery peptides and discuss important related factors such as NP–peptide bioconjugation, uptake efficiency, intracellular fate and toxicity. We also highlight various demonstrations of therapeutic applications of NP–peptide conjugates where appropriate. The paper concludes with a brief forward-looking perspective discussing what can be expected as this field develops in the coming years.
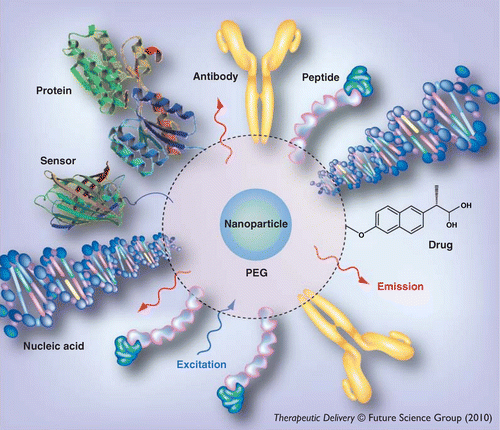
Multifunctional nanoparticle assembly. Shown is a representative nanoparticle decorated with multiple disparate functional molecules (e.g., nucleic acids, proteins, drugs and peptides). Robust conjugation of biomolecules to the nanoparticle surface is critical for the development of ‘value-added’ constructs that can serve multiple functions within one nanoparticle platform.
PEG: Polyethylene glycol.

(A) Electrostatic interactions use opposite charges on the surface of the NP and the peptide to mediate charge-charge-based NP–peptide assembly. (B) Direct interaction involves certain peptide motifs that can bind to/coordinate with the NP surface with high affinity. Examples include the interaction of free thiols with the surface of Au-NPs and the high-affinity coordination of polyhistidine tracts with NPs (e.g., QDs) with Zn2+-bearing surfaces. (C) Secondary interactions utilize specific ligand-receptor interactions and are almost completely exemplified by the biotin–streptavidin interacting pair. The incorporation of the biotin moiety at the peptide’s terminus can mediate directional assembly of the peptide with the nanoparticle. (D) Covalent attachment linkages utilize classical bioconjugation chemistry such as EDC-based coupling of amines to carboxyls and NHS- and maleimide-mediated conjugation to amines and thiols.
Bt: Biotinylate; EDC: 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide hydrochloride;
NHS: N-hydroxysuccinimide.

(A) Polyarginine-mediated delivery of green-emitting CdSe/ZnS quantum dots (QDs) to human embryonic kidney cells (HEK 293T/17). Endosomes are counterstained with AlexaFluor-647-labeled transferrin. The inset shows the colocalization of the QDs within endocytic vesicles. (B) Live cell imaging of 655 nm-emitting streptavidin-conjugated QDs delivered via TAT peptide to HeLa cells. Red arrows show QD-loaded vesicles being trafficked along filipodia and the red box highlights a vesicle that was recently shed to the extracellular medium. (C)In vivo tissue imaging using QD-TAT-labeled mesenchymal stem cells. Shown are red-emitting QDs (arrow) contained within mesenchymal stem cells that localized to the lung after injection into mouse tail vein. (D) TAT-based ‘smart’ micellar pH-responsive drug delivery system. TAT peptides are ‘shielded’ at physiological pH by electrostatic interaction with polysulfonamide moieties and become exposed at lower pH (tumor environment). (E) pH-dependent uptake of the TAT-micellar delivery vehicle shown in (D). In MCF-7 cells at pH 7.4 (left, TAT shielded) no uptake is seen while robust uptake occurs at pH 6.6 (right). (F) Preferential expression of luciferase in the rat cervical spinal after intrathecal injection of commercial magnetic NPs complexed with PEI and native TAT. Application of an external magnetic field (right panel) concentrated transgene expression in the meninges (arrows) of the spinal cord relative to absence of applied magnetic field (left).
PEG: Polyethylene glycol; PEI: Polythelenimine; PLLA: Poly(l-lactic acid).
(A & C) Modified with permission from [35,70] © ACS.
(B) Reproduced with permission from [69] © ACS.
(D) Reproduced with permission from [7] © Elsevier Publishers.
(E) Modified with permission from [7] © Elsevier Publishers.
(F) Modified with permission from [74] © Elsevier Publishers.

(A) Arginine–glycine–aspartic acid (RGD)-functionalized CdSe/ZnS QDs for labeling osteoblasts. Efficient labeling was dependent on spacer length. (B)In vivo near-infrared imaging of tumor-bearing mice (left shoulder, white arrows) using RGD-conjugated 705 nm-emitting quantum dots (QDs). QDs accumulated at the tumor site in the animal to the left (QD-RGD) while they did not in the animal to the right (QD only). (C) Confocal scanning microscopy of cellular uptake of doxorubicin-poly(lactic-co-glycolic acid)-RGD NPs in B16F10 cells overexpressing αvβ3 integrins on their membrane. Note the perinuclear morphology of the NPs.
(A) Reproduced with permission from [80] © Wiley-VCH Publishers.
(B) Reproduced with permission from [85] © American Chemical Society.
(C) Reproduced with permission from [96] © Springer Science Publishers.

(A) Pep-1-mediated delivery of lysosome-specific antibody. Fluorescein isothiocyanate-conjugated anti-Lamp-1 antibody was delivered to COS-7 cells by complexing with Pep-1. (B) Pep-1-mediated delivery of quantum dots (QDs) for drug screening. CHO cells expressing the receptors muscarinic (M1, orange circle) or serotonin 2A (Ser 2A, yellow circle) were encoded with 608 nm and 582 nm QDs via Pep-1 delivery, respectively, mixed, seeded to microtiter plates and loaded with a calcium-responsive dye (left panel). Only those cells expressing the M1 receptor displayed increased fluorescence when stimulated with carbachol (right panel). (C) Induction of apoptosis by GH3 peptide-decorated QDs delivered by Pep-1. Red-emitting QDs are localized in the nucleus of HeLa cells. Nuclear condensation and fragmentation (arrow) and membrane blebbing (asterisk) of apoptotic nuclei are indicated. (D) Effect of transduced Pep-1-HSP-27 on neuronal cell viability after ischemic insult in the gerbil hippocampus. Photomicrograph shows the robust cresyl violet staining of gerbil hippocampus 7 days after ischemic insult in gerbils injected with Pep-1-HSP-27 (lower panel) compared with vehicle control (upper panel).
(A)Reproduced with permission from [104] © Nature Publishing Group.
(B) Modified with permission from [110] © Elsevier Publishers.
(C) Reproduced with permission from [25] © The Royal Society of Chemistry.
(D) Modified with permission from [112] © Wiley-Blackwell Publishers.

(A) 3 nm fluorescent Au-NPs (blue) accumulated in the cytoplasm and nucleus of HeLa cells. Nuclei are counterstained with SYTO 59 (red). (B) Allatostatin 1 (Ast 1)-streptavidin QDs delivered to NIH 3T3 cells. Ast 1-QD conjugates were localized to the cytoplasm and nucleus after endocytosis. (C) Localization of intravenously administered luciferase plasmid DNA/poly (amido amine)–polyethylene glycol NPs to mouse brain.
(A & C) Reproduced with permission from [116,125] © American Chemical Society.
(B) Reproduced with permission from [122] © Royal Society of Chemistry.
Nanoparticles in therapeutic applications
▪ Therapeutic delivery & the role of nanoparticles
The goal of pharmaceutical research has traditionally focused on the development of new drug formulations and novel therapeutic compounds to treat an array of diseases. Coupled with this pursuit is the need to develop effective delivery modalities. While many drugs have successfully entered the commercial market, progress along this pipeline is not without limitations. For example, many drugs are compatible with only a limited number of delivery methods and are typically designed for systemic delivery, where they are susceptible to metabolic breakdown [1–3]. Furthermore, systemic delivery requires high dosage levels as the drugs are distributed and partitioned throughout the body. As a result, systemically administered drugs are often hampered by nonspecific toxicity and side effects in nontargeted cells and tissues, often limiting the number of doses that can be administered to patients. Thus, the need to identify more specific and targeted delivery modalities to increase the therapeutic indexes of drugs remains a key roadblock in the development of the next generation of therapeutics [4]. It is against this backdrop that the burgeoning field of nanoparticle (NP)-mediated drug delivery (NMDD) has emerged, to potentially address a number of the critical issues facing the delivery of pharmaceuticals.
Nanoparticles encompass a class of materials broadly defined as being 100 nm or less in size and span an array of compositions including metals, semiconductor quantum dots (QDs), oxides, polymers, vesicles (e.g., micelles/liposomes), carbon-based materials (e.g., nanotubes, fullerenes and nanodiamonds) and protein- and nucleic acid-based particles. Examples of these materials and some of their unique properties and potential therapeutic properties are listed in Table 1. NPs also possess a number of physical attributes that make them attractive for use in therapeutic and biomedical applications. Their small size allows them to gain access to areas that are otherwise not reachable by other materials (e.g., the blood–brain barrier [BBB], the CNS, the GI tract, capillaries and the lymphatic system). Second, their high surface-to-volume ratio affords them the ability to be decorated with a large cargo ‘payload’ on a relatively small frame; large numbers of cargo molecules (e.g., drugs and labels) can be loaded onto just a few particles. Perhaps the concept driving the most interest in this area is the potential role of NPs in the development of ‘theranostic’ materials; materials that incorporate both a diagnostic and a therapeutic capability into a single species. This arises from NPs’ unique size combined with their ability to be ‘loaded’ with multiple disparate functional moieties (Figure 1). When these NPs are then further conjugated to small targeting peptides (typically <40 amino acids), ‘value-added’ constructs that are capable of far more than each individual component can be realized. Select examples from the literature have already demonstrated the feasibility of generating hybrid NP–peptide constructs in which the peptide adds a critical new function not inherently possessed by the NP (e.g., sensing and homing to cells). For example, micelles and liposomes that are functionalized with cell-penetrating peptides (CPPs) derived from the HIV-1 Tat protein yield hybrid ‘smart’ materials that can respond to the acidic pH environment of the tumor interstitium, allowing for cellular uptake of the drug-loaded vesicles only after they are appropriately targeted. This is achieved by the ‘shielding’ of the CPP with either a pH-sensitive binding partner or by burying the CPP within a pH-sensitive layer of poly(ethylene glycol) (PEG) [5–8]. In this review we examine the current trends in the use of peptides for the cellular delivery of a range of NP materials of biomedical relevance. We begin with a brief discussion of the various methods for the delivery of NPs to cells and current bioconjugation techniques that are available for generating functional NP–peptide constructs. We then present recent examples from the literature demonstrating the cellular delivery of a variety of NPs using the most commonly employed peptides. Both in vitro and in vivo examples of the peptide-mediated delivery of NP-based imaging agents (fluorescence and magnetic resonance), drug-delivery vehicles, therapeutic proteins and nucleic acids are highlighted and, where appropriate, the therapeutic applications of these constructs are presented.
▪ Cellular delivery of nanoparticles
Several features are desirable in the development of delivery strategies for the intracellular delivery of NPs for therapeutic purposes. These include:
▪ The ability to deliver sufficient amounts of NP materials intracellularly to mediate the desired function (e.g., cellular labeling/imaging, sensing and drug delivery);
▪ The ability to deliver the NPs in a specific, controlled manner to only the targeted cell population;
▪ The elicitation of minimal cytotoxicity.
Currently, the approaches available for the intracellular delivery of NPs can be broadly grouped into three main strategies based on their inherent physicochemical properties.
Passive delivery relies on the use of the inherent physicochemical properties of the NP itself (e.g., surface composition, functionalization and charge) to mediate cellular uptake. The uptake process is carried out by pinocytosis, a nonspecific, nonreceptor-based form of endocytosis in which minute amounts of liquids, solutes and small materials are internalized within plasma membrane-derived vesicles. This delivery approach has been used for the labeling of numerous cell lines with a variety of NP materials including nanodiamonds [9], QDs [10] and Au-NPs [11]. While this delivery approach is noninvasive and requires no functionalization of the NP surface other than to render the NP hydrophilic/soluble, it lacks the degree of specificity or cell-targeting ability that is required for therapeutic applications. Passive delivery also typically requires that a large amount of material be exposed to the cells to compensate for the lack of specific uptake.
Active delivery involves the direct physical manipulation of the cell (specifically, the plasma membrane) to introduce NPs into the cell. Techniques such as electroporation and nucleofection, originally developed for the cellular delivery of nucleic acids, utilize a brief electrical pulse to permeabilize the phospholipid bilayer of the plasma membrane to allow entry of membrane-proximal materials into the cellular cytosol. Nucleofection further utilizes a transfection reagent to direct the delivered materials to the nucleus. These techniques have been demonstrated for a variety of NP materials, including QDs [12], silver NPs [13], poly(lactic-co-glycolic acid) (PLGA) NPs [14] and various nucleic acids [15]. Microinjection, as the name implies, involves the use of a capillary (typically borosilicate glass) bearing a nanometer-sized orifice to directly deliver femtoliter amounts of materials to the cytoplasm [16,17]. Although they have the ability to deliver large amounts of NP material directly to the cellular cytosol, active delivery methods are limited in their therapeutic utility in several ways. Electroporation and nucleofection are exclusively in vitro techniques and are not amenable for in vivo use, . Furthermore, they lack specificity and are often associated with a high degree of cell mortality. Microinjection, while highly specific, is limited to the manipulation of only a few cells at a time and, thus, is limited in its throughput.
Facilitated delivery involves the decoration of the NP surface with biological (e.g., peptides, proteins or nucleic acids) or chemical (e.g., lipid-based transfection reagents, drugs or small nutrients) moieties aimed at targeting specific cell surface receptors. Within this group, peptides offer several unique advantages that make them rather attractive for therapeutic delivery applications. Their small size minimizes the overall radius of the resulting peptide–NP conjugate while still affording a high valence (number of peptides per NP). Furthermore, their size reduces immunogenicity in vivo. Peptides are economical and facile to produce as they can be easily synthesized commercially or expressed recombinantly in the laboratory. From a functional perspective, peptides are biocompatible, derived from naturally occurring protein precursors and can be very specific and bind with high affinity to their cognate receptors, often with affinities comparable to those of full-length antibodies [18]. Finally, multiple different peptide species can be arrayed around the NP to incorporate multifunctionality or produce a ‘value-added’ material that serves multiple purposes in one NP. For these reasons, peptides represent a very attractive and useful class of molecules for the development of NPs for therapeutic delivery.
Bioconjugation of peptides to nanoparticles
Beyond the individual properties of the NPs and peptides to be exploited in a bioconjugate, perhaps the next most relevant issue is the chemistry utilized to join them together as this has direct ramifications for subsequent function. Before briefly discussing the currently available NP biofunctionalization chemistries, it is quite helpful to visualize six ideal criteria or properties desired from such bioconjugations as they illuminate the potential impact on final utility [19]. Ideally, the chemistry would attach the peptide to a NP in one of the following ways:
▪ In a homogenous manner;
▪ With control over its final orientation;
▪ With control over its distance from the NP surface;
▪ With control over ratio or valence on the NP surface.
The goal is to uniformly display the peptides on the NP surface with their active regions all clearly extended away and available for activity. For example, the commonly utilized oligoarginine (Argn) motifs used in many CPPs require the positive charge on the arginines to be clearly available for direct interaction with the extracellular membranes, as this initial binding facilitates subsequent endocytic uptake [20]. If the Argn are not clearly available by either being bound around the NP surface, sterically blocked by NP surface ligands or alternatively attached in a heterogeneous manner, this can result in mixed functional avidity with some NPs undergoing high-efficiency uptake while others do not. Further, the density or ratio of CPP on NP surfaces is relevant, as efficient uptake of CPP-functionalized NPs also requires a high ratio of peptide on the surface. Low functionalization levels may not be sufficient for uptake, especially when applying small concentrations of NP materials to cells [20]. Another factor with ramifications, in particular for developing NMDD, is that of controlling the peptides affinity to the NP surface. It is becoming increasingly desirable to attach peptides functionalized with cargos such as drugs, contrast agents, siRNA or sensors to NPs in a manner that allows them to be released intracellularly at a specific location where they can have the most impact. This can only be achieved with a linkage that is responsive to external stimuli (e.g., light or heat) or environmental cues (e.g., changes in pH or presence of a reducing agent/protease) [21]. Last, and perhaps equally importantly, it would be extremely utile if these chemistries would allow any peptide sequence to be attached to any NP material while still maintaining the previously described levels of control [19].
A variety of factors go into choosing which strategy to use when joining a peptide to a NP. These will be dictated primarily by a combination of the NP materials themselves, their characteristics, the ligands on their surfaces along with their functional groups, the peptide sequence itself and the final utility desired. For example, if a particular sequence or functional group is required for subsequent peptidyl activity in the conjugate, such as the presence of a primary amine, then chemistries that specifically target and modify this group would not be favored to yield functional composites. A number of related reviews and sources are available that discuss some of these pertinent issues and chemistries [22–24]. There are currently four general strategies commonly applied for attaching peptides to NP materials (Figures 1 & 2) and each is characterized by its own inherent set of benefits and liabilities [22]. These conjugation strategies include the following.
▪ Electrostatic interactions
This is perhaps the easiest strategy to implement as it usually involves simply mixing the NP and peptide together to facilitate their self-assembly. The NP must present the requisite positively/negatively charged surface with the peptide having the opposite characteristics. Naik and co-workers showed that this approach could even be used to deliver QDs to cells [25]. The positively charged lysine-rich 21-residue Pep-1 peptidyl sequence was noncovalently associated with commercial streptavidin-conjugated CdSe/ZnS core/shell QDs, which facilitated their delivery to HeLa cells. A subsequent mixed-surface approach combined biotinylated peptides with the electrostatically associated Pep-1 to achieve either organelle targeting or apoptotic triggering and highlighted the versatility of this approach. The authors suggested that the Pep-1 carrier initially associated with the QD conjugates to allow initial interactions and crossing of the cell membrane and then dissociated from the complex, thus permitting further intracellular targeting [25]. Electrostatic chemistry has also been applied to negatively charged citrate-stabilized Au-NPs partnered with a positively charged coiled peptide and pH changes were even used as a trigger to alter the electrostatics and control assembly kinetics [26]. The ratio of peptide per NP may be somewhat controlled with this chemistry assuming that all the peptides would fit around the NP, however, it is difficult to control the final orientation that the peptide assumes.
▪ Direct interactions
Depending upon NP material and its surface exposure or access to the surrounding aqueous environment, certain peptidyl motifs can bind directly to NPs via high-affinity interactions/coordination providing access to another means of bioconjugation. The most common binding used in this class is that of thiols directly to Au-NP surfaces and this interaction is extensively described in several reviews [27,28]. As cysteines can be placed anywhere within a peptide sequence, the number and position of thiols can be quite effectively controlled. A further benefit of working with Au-NPs is that should the peptidyl-thiols be present as protected or linked-dithiols, the Au can effectively open/reduce then during attachment. This chemistry is equally applicable with a number of other noble metal NPs including Pt, Pd and Ag. Similar approaches can also be used to attach peptides to the surface of ZnS-overcoated QDs. In this case, the peptides must be reduced first and usually display multiple thiols for higher affinity binding. The process for binding thiols directly to QDs proceeds by a mass action-driven replacement of the native surface ligands and is commonly referred to as ‘cap-exchange’. Several impressive examples demonstrating either intracellular delivery of QDs [29] or single molecule QD-protein tracking [30] have been reported using QD samples prepared with such multi-cysteine phytochelatin peptidyl motifs.
Metal-affinity coordination between polyhistidine residues (Hisn) usually placed at peptide termini and transition metals including, in particular, Zn2+ present on the QD surface has been extensively exploited for assembling a variety of peptides onto QDs. Hisn sequences were originally recombinantly appended to expressed proteins to facilitate their purification over nitriloacetic acid (NTA) media [31]. This facile self-assembly process requires only mixing of the QDs with the Hisn-appended peptide (commonly a His6) to create the final bioconjugate. QD to Hisn-peptide interactions are characterized by rapid assembly (min), with high-affinity equilibrium binding constants measured for solution self-assembly (Kd-1 ∼109 M-1), allowing control over the valence of peptides displayed per QD through modulation of the molar ratios used and even, in most cases, providing control over the peptide’s spatial orientation on the QD surface [32,33]. An ancillary benefit of using Hisn-modified peptide sequences is that they can be collected and purified if required using NTA media [34]. Using this coordination interaction, peptides have been assembled to QDs to create sensors for monitoring proteolytic activity [33], for CPP-facilitated cellular delivery [35] and for co-delivery of QDs displaying CPP along with other fluorescent protein cargos [36]. The principle benefit of using cysteine thiols or Hisn motifs for direct peptidyl binding to QDs and Au-NPs is that they are natural residues and can be easily incorporated into any nascent sequence during synthesis. A similar peptide-based attachment strategy targeting NPs with oxide surfaces is another possibility. Rather than exploiting existing residues, however, it requires that the peptide be modified to display a hydroquinone moiety to facilitate the NP surface binding [37]. Lastly, it is worth mentioning that considerable research has been focused on selecting peptidyl sequences that bind to various metallic surfaces using phage display and related techniques [38,39]. This may soon translate into the availability of still further peptidyl sequences capable of mediating direct interactions with NPs made from the same materials.
▪ Secondary interactions
This class of binding is exemplified and almost totally dominated by biotin–avidin chemistry [23]. The combination of high intrinsic affinity, facile synthetic insertion of biotin groups into peptide sequences as desired, along with commercial availability of Au-NP, QD and other NP materials functionalized with either biotin or avidin/streptavidin also make it one of the most relied upon NP–peptide bioconjugation approaches. An additional benefit is derived from inserting a single biotin into the target peptide, thus preventing any potential cross-linked structures. Placing the biotin at the peptide termini may also provide some control over orientation on the NP. Almost all examples of this chemistry utilize avidin-functionalized NPs and mono-biotinylated peptides. The converse approach is far more complex, as using a tetravalent avidin intermediary with biotinylated NP materials often results in cross-linked aggregates and requires the further step of linking a peptide to the avidin. One further issue to be considered with this chemistry is that the average number of avidin binding sites provided per NP can fluctuate rather dramatically. For example, commercially available streptavidin-functionalized QDs are loosely characterized as displaying 5–15 streptavidin molecules each with three nominally available biotin-binding sites, yielding an estimated maximum of 15–45 conjugation sites per NP [201]. This suggests that quite a bit of heterogeneity in peptide valence may be present across the final NP–bioconjugate ensemble sample.
There has also been a concerted effort to develop NTA ligands for NP surface functionalization, as these groups can substantially expand NP–bioconjugation access by providing targeted binding to Hisn sequences beyond the direct QD coordination, as described previously. Although the NTA groups are first chemically immobilized onto the NPs surface ligands, the binding to peptide is indirect or secondary in this context. The direct benefit of this chemistry is that potentially any peptide containing a Hisn sequence can be conjugated to any NP functionalized with an NTA group and examples of NP materials thus functionalized to date include Au-NPs [40]; FePt magnetic NPs [37], semiconductor QDs [41], Ni-NPs [42] and Si-NPs [43]. NTA functionalized NPs, primarily Au (nanoprobes), are also available commercially along with some reactive NTA groups.
▪ Covalent linkage to surface ligands
Following biotin–avidin assembly, the next most popular coupling strategy covalently links the peptide to the NP’s surface functionalization ligands; the latter serve to primarily provide solubility but may also prevent nonspecific interactions. In most cases, these linkages exploit ‘classical’ bioconjugation chemistry to complete the composite and target primarily amines – thiols and carboxyl groups – as these are most common on both peptides and NPs and are easily amenable to modification [23]. Chemistries utilized include 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide hydrochloride (EDC)/N-hydroxysuccinimide (NHS) to join amines to carboxyls, maleimides which target reduced thiols, along with thiol-exchange, which also targets thiols [23,44]. The choice in chemistry and groups to be linked will be driven by what is present on the termini of the NP surface ligands and what can be targeted on a peptide without compromising activity. The single best resource on these types of chemistries is Hermanson’s Bioconjugate Techniques, which provides an almost exhaustive overview of all possibilities [23]. Although focused on modifying biomolecules, the discussion is still directly relevant as almost all NP-modification chemistries are drawn from those applications. Another resource is the scientific literature itself, as the examples where this type of chemistry have been applied are too numerous to list. Again some NP materials (mostly Au-NPs, Nanoprobes Inc., NY, USA) are commercially available prefunctionalized with reactive NHS-esters and maleimides targeting primary amines and thiols, respectively. Several other chemical strategies are currently being developed to accomplish the same types of NP conjugations, including copper (I)-catalyzed azide–alkyne cycloaddition (CuAAC), chemoselective ligation along with several enzyme-catalyzed modifications [45–48]. The potential benefit of these latter chemistries is derived from their bio-orthogonality. That is they join two unique cognate groups together without modifying any other functional groups already present on either the NP or peptide.
Several other factors should also be considered when bioconjugating peptides to NPs. In many instances, the final NP–peptide conjugate must be purified from unreacted reagents and byproducts. This can be either quite simple or rather challenging depending upon materials and conjugate properties. For example, separating semiconductor QD–peptide conjugates (MW in situ 100 kDa) from free peptides (MW < 5 kDa) can be readily accomplished with a desalting column [46]. By contrast, purifying Au-NPs that are monolabeled with a peptide from unlabeled Au-NPs can require extensive optimization with different separation techniques and chromatographic materials [49]. There are many techniques that can be applied for these purposes, including chemical extractions, electrophoresis, various types of chromatography (i.e., HPLC and LC) and differential centrifugation. However, beyond the protocols sometimes provided by NP manufacturers, there are few direct resources to draw on besides examples reported in the literature. Other issues that may be relevant include the overall size of the final bioconjugated NP and whether this plays any role on the subsequent application. For example, NP–peptide conjugates with large hydrodynamic radii may be incompatible with certain functions, such as nuclear uptake, due to nuclear pore size limitations. Overall conjugate yield, the number of preparatory steps and the ease of preparation should also be carefully considered, such that adequate materials are ready when required. Furthermore, as individual NP–peptide pairings will be physiochemically disparate from other composite materials and may also be intended for different applications, each should be designed and assembled independently while considering the issues iterated here. A final consideration that warrants mentioning is the adsorption of nonspecific species (e.g., proteins or peptides) to the NP surface in biological systems. A number of approaches have been taken to passivate the NP surface including, for example, the use of PEG chains to simultaneously impart water solubility and sterically block the adsorption of nonspecific proteins.
Peptides in the therapeutic delivery of nanoparticles
In this section we highlight some of the most commonly used peptides that have been employed for the cellular delivery of NPs and their therapeutic applications are discussed where appropriate. Each peptide and the associated NPs for which it has been used for cellular delivery are summarized in Table 2.
▪ TAT & TAT-like peptides
The TAT peptide was one of the first cell-penetrating peptides (CPPs) to be described. Interest in this peptide stemmed from two independent reports demonstrating that the 86-residue transcriptional activator protein Tat (encoded by HIV-1) could be efficiently internalized by cells when present in the surrounding tissue culture media [50,51]. Subsequent studies to determine the domain responsible for cellular uptake localized the activity to residues 47–57 within the native Tat protein [52]. The corresponding 11-mer peptide sequence (YGRKKRRQRRR) bears six arginines and two lysine residues and these positively charged residues have been identified as the key determinants of cellular uptake as they mediate the initial interactions of the peptide with the negatively charged cell surface [53–56]. Accordingly, peptides based on repeats of or domains rich in positive residues (e.g., polyarginine and polylysine) have found great utility as mediators of NP cell internalization [57–60]. While it has been firmly established that the positively charged residues are responsible for uptake, the exact mechanism of cellular uptake remains somewhat less clear. Indeed, a number of mechanisms for the uptake of TAT and TAT-like peptides and their associated cargos have been proposed. Evidence exists for two types of endocytosis: classical clathrin-mediated endocytic uptake and caveolae-mediated clathrin-independent endocytosis. The work of Console et al. supported clathrin-mediated uptake as the authors showed that the TAT peptide colocalized rapidly (within 1 h) with the classical clathrin-mediated endocytosis marker transferrin within endosomes [61]. Further corroborating this, Lundberg et al. showed that the uptake was inhibited when the delivery was performed at 4°C, which inhibits endocytosis [62]. Other evidence points to caveolae-mediated endocytosis involving the formation of membrane invaginations composed chiefly of cholesterol and sphingolipids. Referred to as lipid rafts, these invaginations mediate a clathrin-independent internalization and are several hours slower than clathrin-mediated endocytosis (reviewed elsewhere [63]). Regardless of the ultimate mechanism of internalization, it is clear that electrostatic interactions between the positively charged residues of the TAT peptide and the negatively charged residues of heparan sulfate proteoglycans (and other cell surface receptors) are essential for initial membrane binding followed by uptake [55,64]. Typically the endocytosed NPs remain sequestered within endocytic vesicles [35,65], although in some instances nuclear entry has been noted [66,67]. The TAT peptide and its derivatives have been used for the cellular delivery of a variety of NPs with implications for therapeutic applications in many cases.
Cellular labeling & imaging
TAT peptides have found great utility as the basis of cellular fluorescence and magnetic labeling reagents in conjunction with various NPs, and both in vitro and in vivo examples have been reported. Fluorescence labeling studies using QDs have shown the importance of tracking the fate of the TAT-delivered materials as NP intracellular fate can vary amongst different materials [63]. We used an oligoarginine-containing TAT-like peptide (R8) to controllably deliver CdSe/ZnS core/shell QDs into HEK 293T/17 and COS-1 cells [35]. Self-assembly of the peptide onto the QD surface was driven by metal affinity interactions between the peptide’s polyhistidine tract and zinc ions on the QD surface. QD uptake was highly specific for the presence of the peptide and the efficiency of internalization tracked with both QD concentration and the number of peptides assembled onto the QD. The QDs remained sequestered within endosomes (Figure 3A) and no localization of the QDs to the nucleus was observed, even after fixation with paraformaldehyde (an experimental artifact that has been reported for TAT-delivered fluorophores and other small molecules [68]). Importantly, minimal inhibition of cellular proliferation was noted in both cell lines tested at the concentrations (∼50–75 nM QD) and incubation time (∼1 h) required for efficient labeling, demonstrating the biocompatibility of the NP–peptide conjugates. We did, however, note that higher concentrations and longer incubation times resulted in considerable inhibition of cellular proliferation. In a subsequent study, this same QD–CPP construct was decorated with fluorescent proteins of varying sizes, demonstrating the utility of the bioconjugate as a platform for the cellular delivery of associated protein cargoes. Proteins as large as 1000 kDa could be efficiently delivered using this approach [36]. Ruan and co-workers assembled a biotinylated form of the native TAT peptide sequence onto commercial streptavidin-coated QDs and monitored the translocation of QD-loaded endocytic vesicles along the microtubule network in live HeLa cells [69]. In addition to showing the accumulation of the QDs within the microtubule-organizing center (perinuclear spaces), the authors also demonstrated the previously unreported exocytosis of large QD-loaded vesicles from the tips of filopodia by vesicle shedding (Figure 3B). TAT has also been used for tracking the intracellular fate of Au-NPs. de la Fuente et al. demonstrated efficient TAT-mediated delivery of Au-NPs to human fibroblasts and showed that intracellular fate was dependent on NP size; 5 nm Au-NPs gained entry into the nucleus while 30 nm Au-NPs remained in the cytoplasm [66,67]. This finding has also been corraborated by other groups [65].
In vivo fate tracking
Cellular labeling with TAT–NP bioconjugates has been extended by a number of groups to include in vivo fate tracking. Lei et al. covalently conjugated native TAT peptide to CdSe/ZnS QDs to label mesenchymal stem cells that were subsequently injected into the tail veins of nude mice [70]. After organ harvesting, the fate of the grafted cells was tracked by fluorescence primarily to the lung, liver and spleen with little or no QD accumulation in the brain, heart or kidney (Figure 3C). The Weissleder group conjugated the native TAT peptide to superparamagnetic iron oxide NPs for the efficient labeling of human CD34+ hematopoietic cells where, despite their overall size of approximately 45 nm, the conjugates were found in both the cytoplasm and the nucleus and had no deleterious effect on cell viability, differentiation or proliferation [71]. Upon injection into nude mice, a percentage of the labeled cells homed to bone marrow and could be recovered by magnetic separation techniques, demonstrating the potential of such an application for use in the analysis of stem cell–organ interaction in stem cell therapy.
Drug delivery
TAT peptide has also been used effectively for the cellular targeting of drug carriers and their associated cargos. Sethuraman and co-workers have described a ‘smart’ micelle-based system wherein TAT was used to target pH-responsive drug-carrying vehicles to acidic solid tumors [7,8]. The delivery construct consisted of 1-a drug-loadable core made of poly(l-lactic acid) (PLLA) surrounded by a hydrophilic PEG conjugated to the TAT peptide and 2-a pH-sensitive copolymer of methacryloyl sulfadimethoxine (PSD) and PEG. At physiological pH, the PSD remained electrostatically complexed with the TAT peptide, blocking TAT’s cell uptake ability. Under acidic pH conditions (e.g., in the proximity of a tumor), the PSD dissociated and allowed the TAT peptide to mediate cellular uptake. The pH sensitivity of this ‘shielded’ delivery vehicle was tested in vitro in MCF-7 breast cancer cells where effective cellular entry and localization within the nucleus was observed at pH 6.6 while no uptake of the drug vehicle was observed at pH 7.4 (Figure 3D). This work was further extended to show the pH-dependent specific uptake and resulting cytotoxicity of the anticancer drug doxorubicin in MCF-7 cells [8]. Another pH-responsive drug delivery NP construct utilizing TAT has been described wherein long, acid-hydrolyzable PEG chains that ‘shield’ a hidden native TAT peptide on the surface of liposomes are cleaved in the low pH tumor environment, thus, exposing the TAT peptide to mediate the liposome drug vehicle [5,6]. Liu et al. formulated PEGylated liposome NPs loaded with an iron core that were conjugated to TAT and shown to cross the blood–spinal cord barrier (BSCB) in a rat model of spinal cord injury, demonstrating the potential utility of the NPs for imaging the injury site [72].
Nucleic acid delivery
TAT and TAT-like peptides have also been employed for the cellular delivery of therapeutic nucleic acids. Han and co-workers functionalized bacterial magnetic nanoparticles with poly (amido amine) (PAMAM) and native TAT peptides and complexed them with siRNAs specific for the downregulation of human EGF receptor (EGFR), which is often overexpressed in many cancers. Effective complex uptake, suppression of EGFR expression and inhibition of tumor cell proliferation and invasiveness was demonstrated in glioblastoma U251-MG cells in vitro and in vivo in a nude mouse model [73]. Song et al. constructed ternary NPs consisting of polyethylenimine-coated magnetic iron beads that were noncovalently decorated with native TAT peptides and plasmid DNA encoding a luciferase reporter construct. Both in vitro (human NT2 neural stem cells) and in vivo (rat spinal cord injection), the presence of the TAT peptide increased gene expression fourfold [74]. Furthermore, the magnetofection complexes in the cerebrospinal fluid responded to a moving magnetic field; shifting away from the injection site and mediating transgene expression in a remote region (Figure 3F). This type of combinatorial approach has implications for the development of TAT-mediated targeted gene therapies that are controllable in vivo.
▪ RGD peptide
The short RGD peptide domain was first identified as the cell recognition sequence in the extracellular matrix (ECM) protein fibronectin [75]. Later, more ECM proteins, including vitronectin, fibrinogen, collagen, laminin and osteopontin were found to contain this sequence in their cell-recognition domains. These ECM proteins interact with a subset of the integrin superfamily of cell surface receptors. These receptors contain α and β subunits that combine in a pairwise manner to bind to different ECM proteins. The binding of integrins to ECM proteins mediates cell–cell and cell–substratum interactions and is involved in the regulation of such cellular homeostasis functions as cell growth, motility, differentiation and angiogenesis [76]. As a result, aberrant expression of RGD-ligating integrins has been implicated in a number of pathological conditions ranging from the platelet response to a variety of cancers [77,78]. Thus, there has been much interest in the development of imaging reagents and therapeutics aimed at exploiting the important role of this cell-binding motif and, to date, RGD-functionalized NPs targeting integrins have been used for cell and diagnostic imaging, drug delivery, gene therapy, orthopedic implants, tissue engineering and various multimodal combinations of the above.
Cellular labeling & imaging
The cellular targeting of integrins has been demonstrated with streptavidin-coated CdSe/ZnS core/shell semiconductor QDs conjugated to biotinylated RGD. A cyclized RGDfK (f: D-Phe) peptide was used to target αvβ3 integrins, which are often overexpressed in a variety of malignancies and play a role in tumor metastasis [79–81]. Binding of the biofunctionalized QD to the integrins on osteoblast cells was successful, but was shown to be dependent on a number of factors including peptide specificity, spacer length between the peptide ligand and the QD, ligand concentration and density and surface topography [80]. Spacer length between the peptide and QD was a key determinant in successful labeling. A 1.7-nm spacer showed only weak nonspecific binding, while a 5.7-nm spacer allowed greater access to the deep RGD-binding pocket, resulting in a high degree of specific uptake (Figure 4A). RGD peptides conjugated to other NPs have been used to target αvβ3 integrin-expressing tumor cells both in vitro and in vivo for their utility in diagnostic cancer imaging (Figure 4B)[82–85]. The use of RGD peptides to target tumors with iron oxide particles for MRI and PET imaging has also been demonstrated [83]. Other cellular targeting applications include fluorescent labeling of mesenchymal stem cells [86] and integrin platelets (targeting integrin GPIIb-IIIa) [87]. As αvβ3 integrins are also overexpressed in the endothelial cells that surround tumor blood vessels, they represent viable targets for anti-angiogenic therapy [88]. RGD peptides, therefore, can be useful therapeutics to block the process of angiogenesis, as well as for targeting diagnostic imaging agents to the tumor vasculature. As imaging of the vasculature must be done in vivo, fluorescent imaging applications must utilize emission in the infrared (IR) or near-IR range (700–800 nm). Numerous groups have begun to image the tumor vasculature using near-IR QDs [85,89,90]. Cai et al. showed colocalization of in vivo QD-RGD signal in the tumor with the vascular marker CD31, indicating the bioconjugated QD does not extravasate out of the vasculature [85]. Later, in vivo real-time vascular imaging showed similar results with specific localization of QDs to regions of high levels of αvβ3 integrin expression along the vasculature [89]. Similarly, radiolabeled RGD peptides and RGD-conjugated magnetic nanoparticles have been used for MRI and PET imaging of tumor vasculature in vivo[91–93]. These include Gd-loaded liposomes and 125I-labeled dendritic nanoprobes. Overall, a high amount of specificity is observed for tumor vessels with these compounds, though undesired accumulation is often observed in other organs in animal models. Various other radiolabeled RGD peptides have been created to target angiogenic vessels that could potentially be conjugated to nanoparticles for delivery. This remains an active area of research and a comprehensive review of RGD ligands targeted to angiogenic vessels is provided elsewhere [91–93].
Drug delivery
Owing to the role of integrins in cellular adhesion and potentially in tumor progression and metastasis, RGD peptides themselves can be considered a therapeutic as they block integrin function. Attachment to NPs may improve their pharmacokinetics, for example increasing their half-life [94]. Additionally, given their affinity for tumors, RGD peptides can also be effective delivery vehicles for chemotherapeutic agents, as exemplified by their use to deliver doxorubicin and paclitaxel via PLGA-nanoparticles [95–97]. In cellular delivery experiments the NPs allowed for controlled drug release and an increased affinity to αvβ3 integrin-expressing cells [95–97]. In in vivo mouse models, the drugs showed aggregation and sequestration in the tumor blood vessels and few, if any, NPs were detected within the tumor itself. Nevertheless, modest effects in primary tumor growth and apoptosis were seen in the tumor, particularly in regions directly adjacent to the blood vessels. Increased survival and a significant increase in antimetastatic activity were also observed in treated animals, indicating some degree of successful drug delivery. Importantly, lower drug-induced weight loss was noted for the NP-delivered drugs when compared with systemic delivery [95–97]. A protease-cleavable RGD peptide (iRGD) has also been reported which has an improved tumor extravasation capability. Here, the RGD portion of the peptide targets the tumor endothelium where proteolytic cleavage exposes a binding motif for neuropilin-1, which mediates penetration into tissues and cells. Abraxane®, a nanoparticulate version of paciltaxel, as well as iron oxide for MRI imaging have both been targeted to tumors in vivo using this peptide [98].
Gene delivery
RGD peptides have also been used to target DNA- and siRNA-containing NPs to specific cells or disease sites. Cellular assays have focused primarily on delivering DNA encoding luciferase or fluorescent proteins as model systems to visualize gene delivery [99,100]. The principal roadblock in this approach remains the endosomal sequestration of most NPs after cellular delivery. Several groups have induced endocytosis with reagents such as chloroquine to release the DNA from the endosome [100]. Therefore, current research efforts also include developing improved methods to release the NP-bound DNA from the endosome and into the nucleus for optimal expression. As RGD-mediated gene delivery is still being optimized in cellular applications, in vivo applications are also in their infancy. As with the cellular assays, most early applications have focused on studying the distribution and metabolism of the DNA and NPs in animal models [101] or the delivery of reporter constructs (e.g., luciferase) to visualize the efficiency of gene expression in vivo[102,103]. There have been a limited number of attempts to deliver therapeutic siRNA in animal models. For example, NPs comprised of siRNA and (1-aminoethyl)iminobis[N-(oleicylcysteinylhistinyl-1-aminoethyl)propionamide] (EHCO) were used to target human glioma U87 xenografts in mice with anti-HIF-1a siRNA targeting hypoxia-inducible factor HIF-1 [102,103] and polythelenimine (PEI) NPs were used to target VEGF-2 in N2A neuroblastoma cell mouse xenografts [103]. Both cases showed that the nucleic acid-loaded NP was targeted to the tumor but had only modest therapeutic effects, indicating the need for a further understanding of the combined endosomal escape and nuclear expression of the DNA-NP conjugates.
▪ Pep-1 peptide
Unlike TAT and RGD, the peptide Pep-1 does not occur naturally. Rather, it is a rationally designed synthetic peptide intended to incorporate three different functional domains into a single peptide moiety capable of cellular entry [104]. The 21-residue amphiphilic peptide (KETWWETWWTEWSQPKKKRKV), available commercially as Chariot™ (active motif), consists of a hydrophobic tryptophan-rich domain containing five tryptophan residues (KETWWETWWTEW) to promote interactions with the cell membrane, as well as hydrophobic interactions with other peptides and proteins; a hydrophilic lysine-rich domain (KKKRKV) from the nuclear localization signal (NLS) of simian virus 40 (SV40) large T antigen to improve intracellular delivery and peptide solubility and a spacer sequence (SQP) containing a proline residue to impart flexibility between the two domains. The synthesized peptide is acetylated at its N-terminus and bears a cysteamide group at its C-terminus as both modifications have been shown to be important for peptide stability and efficient cellular uptake [104,105]. Iterative functional studies of the Pep-1 sequence have determined that all four of the cationic lysine residues within the hydrophilic domain are required for optimal membrane interaction and that the presence of the proline within the spacer domain is essential for maintaining the required flexibility between the hydrophobic and hydrophilic domains [106]. Lastly, the Pep-1 peptide adopts a helical structure and the presence of the tryptophan–phenylalanine tandem on one side of the helix is required for cell membrane interaction and membrane disorganization functions [107]. The current understanding of the Pep-1 uptake process is that, unlike many other CPPs, it is independent of endocytosis, as demonstrated by the efficient uptake of Pep-1-cargo complexes even at 4°C [104]. Studies have demonstrated that for most cell lines the optimal Pep-1:protein/peptide ratio for efficient nonendosomal delivery is approximately 20:1 [108] and at higher ratios the delivery becomes partially endosomally sequestered and less efficient, suggesting some overlap with other cellular uptake pathways [107].
Based on structural and biophysical studies, a mechanism has been proposed for the Pep-1-mediated cellular entry of materials that involves formation of the Pep-1-cargo complex; binding of the complex to the cell surface (first involving electrostatic interactions with proteoglycans then with phospholipid head groups); insertion of the complex into the membrane; and release of the Pep-1 cargo complex into the cytosol and targeting to intracellular structures [109]. Pep-1 has found the greatest utility in the areas of intracellular labeling and the delivery of therapeutic proteins.
Cellular labeling, imaging & therapy
The first example of cellular labeling using the Pep-1 peptide came in the seminal report describing the peptide’s cell internalization ability [104]. Morris et al. showed the Pep-1-mediated cellular labeling of numerous cell lines with Pep-1-fluorophore conjugates and Pep-1 complexed with various fluorescent proteins and antibodies. Initially they demonstrated the rapid uptake, in less than 10 min, of fluorescein-labeled Pep-1 into the nucleus of human HS-68 fibroblasts, murine NIH-3T3 fibroblasts and COS-1 cells under standard cell culture conditions with minimal cytotoxicity observed at the concentrations required for efficient labeling. Identical results were obtained when the cells were incubated at 4°C prior to delivery, evidence of a process independent of normal endocytosis. Like TAT, the efficiency of Pep-1-mediated delivery is not affected by the presence of serum (10%), an attractive feature for potential in vivo therapeutic applications. Their work was then extended to the delivery of various proteins spanning a range of sizes: 30 kDa green fluorescent protein, 120 kDa B-Gal and 150 kDa full-length antibodies directed towards specific intracellular structural proteins (β-actin and lysosome-associated membrane protein) (Figure 5A).
Pep-1 has also proven useful for cellular labeling and imaging applications utilizing QDs, with reports demonstrating the use of Pep-1-QD complexes in drug-screening assays, the induction of specific cellular functions/phenotypes and potentially for in vivo cell therapy and tissue engineering. Mattheakis et al. developed a drug screening system based on the Pep-1-mediated delivery of QDs to CHO cells [110]. Three different CHO cell lines, each expressing a different G-protein coupled receptor were uniquely ‘coded’ with a different color-emitting QD (582 nm [serotonin 2A receptor], 608 nm [muscarinic receptor] and 630 nm [serotonin 2B receptor]). Mixtures of the cells were seeded into a microtiter assay plate and loaded with a fluorescent calcium indicator. Upon exposure to a specific agonist of the muscarinic receptor, only those cells QD-coded for muscarinic receptor expression showed a concomitant increase in intracellular calcium fluorescence intensity (Figure 5B). This QD-based barcoding approach, mediated by Pep-1 QD delivery, has applications for multiplexed cell-based drug-discovery assays and, more generally, for studies using mixed cell populations (e.g., tissue engineering). The work of Rozenzhak is another elegant example of a Pep-1-directed QD cell-labeling study with implications for targeted cancer therapy/therapeutic peptide delivery [25]. The authors used commercially available streptavidin-conjugated QDs decorated with both a biotinylated Pep-1 peptide and the pro-apoptotic GH3 peptide to induce apoptosis in vitro. The Pep-1 peptide directed the QDs to the nucleus of HeLa cells and simultaneously allowed for observation of the distinct morphological changes (nuclear condensation and fragmentation) indicative of apoptosis induced by the GH3 peptide cargo (Figure 5C). Studying the internalization of CdSe/ZnS QDs in human adipose-derived adult stem cells (hADAS), Chang et al. observed the effect of the QD delivery route on stem cell function [111]. They noted that, in contrast to lipid-based delivery reagents (which sequestered internalized QDs within the endolysosomal pathway), Pep-1-mediated uptake allowed the QDs to bypass lysosomal degradation. Importantly, no deleterious effects on cellular proliferation or the expression of osteogenic/chondrogenic-specific lineage markers were observed after 28 days in culture. These results highlight the potential of Pep-1 delivered QDs to be used in the tracking of hADAS when employed in cellular therapy and tissue engineering applications.
Delivery of therapeutic proteins
Numerous reports have described the use of Pep-1 for the delivery of therapeutic proteins. In a study of the protective effects of heat shock protein-27 (HSP-27) against ischemia-induced oxidative stress in astrocytes and primary neuronal cells, An et al. took a fusion protein approach [112]. Rather than utilizing the ability of Pep-1 to noncovalently assemble with the therapeutic heat shock protein, they expressed and purified a recombinant Pep-1–HSP-27 fusion protein in bacteria. When added to the tissue culture media of astrocytes and primary neuronal cells, the protein rapidly entered the cells and conferred protection against cell death induced by oxidative stress. Furthermore, when injected intraperitoneally into gerbils prior to ischemic injury, the construct conferred 78 and 70% viability in hippocampal neurons after 4 and 7 days, respectively, relative to untreated control (Figure 5D). Notably, this result demonstrated the ability of the injected therapeutic fusion protein to cross the BBB.
Using Pep-1 in a similar fusion protein scheme, Yune et al. fused Pep-1 to the antioxidant enzyme superoxide dismutase 1 (SOD1) as a therapeutic approach to the detoxification of intracellular reactive oxygen species (ROS) as a result of spinal cord injury [113]. In cultured primary cortical neurons in vitro and in an in vivo rat model of spinal cord injury, the delivered fusion protein significantly decreased levels of ROS and inhibited the activation of the pro-apoptotic enzymes caspase-9 and caspase-3. Zhang et al. used a similar Pep-1-SOD1 fusion construct to impart protection against ROS-induced damage following myocardial ischemic insult in vivo[114]. When injected into the tail veins of Spraque–Dawley rats, the fusion protein significantly improved cardiac function of the left ventricle, decreased infarct size and reduced the release of a number of infarct-associated enzymes.
▪ Organelle-specific peptides
A number of reports have demonstrated the cellular delivery of NPs using peptides aimed at directing the NP cargo to a particular subcellular location or organelle with materials being directed to either the nucleus or the mitochondria. Using this approach, the organelle-specific delivery of QDs, fluorescent Au-QDs, Au-NPs and liposomal-DNA complexes has been realized. These efforts have focused predominantly on in vitro demonstrations, yet they are worth mentioning here as they hold enormous therapeutic potential when coupled with other cellular-targeting capabilities.
In an elegant report, Hoshino et al. described the ability to direct both the uptake and the localization of CdSe/ZnS QDs to either the nucleus (using a nuclear localizing peptide [R11]) or to the mitochondria using the mitochondrial targeting peptide derived from cytochrome C oxidase (MSVLTPLLLRGLTGSARRLPVPRAKIHWLC) [115]. Upon direct conjugation of the peptides to the QDs by coupling via primary amines or free thiols on the QD-capping ligands, a fine level of control was demonstrated in directing the NPs to their respective subcellular compartments. The NLS from the large T antigen of SV40 virus, comprised of the sequence PKKKRKV, has been used for the cellular uptake and nuclear accumulation of a variety of Au-NPs [116–118]. Mandal et al. self-assembled cysteine-terminated NLS peptides onto the surface of 5-nm Au-NPs and tracked their accumulation within the cytoplasm and at the nuclear membrane using transmission electron microscopy (TEM) [118]. Xie and coworkers used surface-enhanced Raman scattering (SERS) to confirm the localization of 20-nm Au-NPs functionalized with NLS peptides within the nucleus of HeLa cells [117}. Using 3-nm fluorescent gold, Lin et al. used a novel ligand-exchange reaction to assemble 11-mercapto-undecanoic acid (MUDA) onto the gold surface followed by conjugation to NLS peptides (Figure 6A). The photoluminescent NPs were clearly observed accumulating in the cytoplasm and the nucleus of HeLa cells [116]. Intracellular delivery of liposome-carrying plasmid DNA has also been reported by a number of groups using nuclear localization signal peptides derived from various sources including SV40 virus [119] and the amino terminal region of the polyoma virus [120].
▪ Neuropeptide
The insect neuropeptide allatostatin 1 (Ast 1) has also been used to deliver NPs to mammalian cells. Ast 1 (APSGAQRLYGFGL) was identified as a member of a family of 13 neuropeptides that are widely distributed in insects and crustaceans whose receptors are highly homologous to somatostatin and galanin receptors [121]. In 2007, Biju et al. identified the peptide’s ability to mediate the rapid uptake of commercial streptavidin-conjugated QDs after its assembly onto the QD via an avidin–biotin interaction linkage [122]. In NIH 3T3 and A431 cells the QDs were endocytosed and localized primarily within the endosomal compartment upon initial uptake. After 1 h, a percentage of the QDs were distributed in the cytoplasm and the nucleus (Figure 6B). More recently, this group identified clathrin-mediated endocytosis as the primary means of uptake. The mechanism by which the QDs are able to escape the endosome and localize to the nucleus, however, remains unclear [123].
▪ Rabies virus-derived peptide
A major obstacle in the treatment of diseases of the neurological system is the presence of the BBB, a series of endothelial cells that precludes the translocation of most molecules (including therapeutics) from the bloodstream to the brain. One possible approach to overcome this issue in the current context is to exploit peptides derived from naturally occurring viruses that have the ability to cross the BBB. In this vein, Kumar and co-workers identified a 29-amino acid peptide derived from the rabies virus glycoprotein (RVG) that enabled the transvascular delivery of a small interfering RNA (siRNA) to the brain [124]. This peptide, named RVG29, bears the sequence TYIWMPENPRPGTPCDIFTNSRGKRASNG. With the knowledge that the peptide bound with high specificity to the acetylcholine receptors (AchR) expressed on neuronal cells, the authors first sought to investigate its utility as a carrier to deliver blood-injected therapeutics to neurons. To mediate the binding of the peptide to siRNA, the authors made a chimeric peptide bearing an oligoarginine (Arg9) domain that bound electrostatically to the nucleic acid. In vitro, the chimeric peptide transfected Neuro-2a but not HeLa cells with siRNA, demonstrating the peptide’s specificity for the AchR and neuronal cells. Furthermore, cell viability was greater than 90%. In vivo, following injection into Balb/c mice of an siRNA targeting the gene encoding Cu-Zn SOD 1, the only organ that demonstrated reduced levels of SOD 1 messenger RNA and protein levels was the brain. The authors then showed the ability of the RVG peptide to noninvasively confer protection via the intravenous delivery of a siRNA targeted against fatal flaviviral encephalitis, wherein the peptide-delivered construct mediated 80% survival compared with controls. Based on the knowledge that the RVG motif could mediate the crossing of the BBB, Liu et al. conjugated the RVG peptide to PEGylated PAMAM dendrimers that were subsequently self-assembled with plasmid DNA encoding create PEG/PAMAM/DNA NPs [125]. Upon intravenous injection, the NPs concentrated in the mouse brain within 80 min (Figure 6C). Gene expression of the delivered luciferase plasmid DNA was significantly higher in all regions of the brain examined relative to control, especially within the hippocampus and substantia nigra. Collectively, these findings show the enormous promise for the development of NMDD-based and noninvasive therapies for the treatment of neurological diseases.
Toxicity of peptide-delivered NPs
A key issue to be addressed is the interaction of the peptides, NPs and NP–peptide conjugates with cells and tissues, and the corresponding toxicity of the delivered materials both in vitro and in vivo. As illustrated herein, the NP materials for which peptide-mediated delivery has been demonstrated to date encompass a range of constituent materials from noble metals, such as gold and semiconductor metals in QDs, to lipid-based materials and full-sized proteins. A thorough characterization of the resulting in vitro and in vivo toxicities is clearly necessary for each newly generated NP–peptide assembly. Over time, studies have evolved from those wherein cells are incubated with rather high NP–peptide concentrations (e.g., 1 µM QDs decorated with mitochondrial-signal peptides [115]) with little regard to cellular toxicity to those in which the minimal amount of NP–peptide conjugate required for efficient labeling coupled with minimal cytotoxicity have been identified [35]. Nonetheless, standard in vitro toxicity studies (typically examining morphology, membrane integrity, cellular proliferation and the induction of oxidative stress) seek to make an initial assessment of the toxicity of the NP, peptide and NP–peptide conjugates in cultured cell lines, which, in most cases, are dedifferentiated, transformed tumor cell lines [126]. While many of the studies presented in this review have demonstrated the biological tolerance of NP–peptide conjugates spanning a range of peptides and constituent materials in cultured cells [35,65,66,69,80,104,110,116] it is important to keep in mind the aberrant nature of these cell lines (overexpression of particular receptors, abnormal chromosome counts) when interpreting the results of cellular uptake and fate. The use of many different materials presenting different surface chemistries in different cell lines can yield disparate results and conclusions. Indeed, some nanotoxicity experts have suggested that these types of stand-alone studies are prejudiced by a lack of thorough material characterization and understanding of the cell lines used [127,128]. Thus, much remains to be learned with respect to the uptake and processing of NP–peptide conjugates [129]. The in vivo toxicity of NP–peptide conjugates is also of paramount importance as their small size allows them to access areas that larger materials cannot (e.g, the respiratory system and tissue interstitium). Accordingly, tissue distribution, clearance rates and the nature of the constituent materials comprising the NP–peptide conjugate become the key determinants of in vivo toxicity and in vivo fate studies have been conducted primarily by following the fate of materials injected into mice or rats [129]. Hence, the need for the careful characterization of new NP materials and NP–peptide conjugates has become increasingly apparent and facilities such as the Nanotechnology Characterization Laboratory of the National Cancer Institute now perform comprehensive evaluations of newly engineered nanomaterials (physical attributes, in vitro biological properties and in vivo compatibility in animal models). Lastly, it is worth noting that in the correct setting (e.g., targeted tumor killing) toxicity is not necessarily an undesirable characteristic and it is expected that future efforts into the understanding of NP–peptide toxicity will focus simultaneously on developing more specific targeting regimes and on tailoring NP toxicity for use in particular applications.
Future perspective
Moving forward, the development of therapeutics based on the peptide-mediated delivery of NPs holds enormous promise. Nonetheless, many challenges remain and ample room exists for improvement on many fronts. Over the next 5–10 years, we anticipate that the field of peptide-mediated NP delivery will see significant advances in the following key areas: bioconjugation methodologies; multifunctionality; cellular targeting and endosomal escape; further characterizations of in vivo toxicity; and significant testing and assessment in animal models. The development of novel and elegant bioconjugation chemistries will be the key driver in mediating the advancements in the subsequent three areas. As described earlier, the attachment of peptides onto the NP surface in a homogeneous manner with control over their orientation, distance and valence is critical for optimal function [19,22–24]. The same holds true for the attachment of any biological, chemical or drug moiety and, as mentioned earlier, there is a growing interest within the field of NMDD to create theranostic NP structures that can effectively combine multiple functions (e.g., cell targeting, imaging and drug delivery). Preliminary examples of such constructs with strong potential for such multifunctionality have already been demonstrated. For example, Duconge et al. developed QDs that were capable of PET imaging by coupling fluorine-18 to phospholipid-encapsulated QD micelles [130]. When injected into mice, these untargeted nuclear-fluorescent particles circulated in the blood before uptake by the reticuloendothelial system and allowed for the quantitative analysis of their in vivo NP distribution. One can envision such a NP being further decorated with a highly specific tumor-homing peptide and a drug moiety to direct the dual-mode imaging NP to a specific tumor site to derive high resolution PET and fluorescence images of the tissue. This approach may even allow imaging and tracking of tumor treatment during the therapy time course. Thus, future work in the area of bioconjugation will seek to develop and refine attachment chemistries that allow for the orthogonal attachment of several disparate biological, chemical or drug species onto the surface of a single NP. The continued isolation of high-affinity targeting peptides is another critical area that will see significant investment in the coming years [18]. The techniques of phage display and combinatorial peptide synthesis/screening will be relied on heavily to identify peptide sequences that can home NP constructs to cells and tissues of choice. When combined with the cellular-uptake peptides described herein, highly specific imaging, sensing and drug-delivery NP conjugates can be realized. Most cellular uptake peptides utilize the endocytic pathway to gain cellular entry. While some peptides have demonstrated the ability to mediate both the uptake and endosomal escape of some NPs (e.g., TAT-Au NPs, Pep-1-QDs), efforts will also need to focus on methods for incorporating an endosomal escape function into those constructs that remain endosomally sequestered. Attempts to achieve this have employed ‘proton sponge’ effect-inducing block copolymers (PEI) [131] or exogenous endo-osmolytic chemicals such as sucrose [132] or chloroquine [133] to induce endosomal release. The accompanying toxicities of these materials, however, warrants further development of endosomal-releasing agents. As improvements are continually made in bioconjugation techniques and solubilization chemistries and further understanding is obtained as to the long-term in vivo fate of the delivered NP constructs, it is anticipated that the first generation of multifunctional NP–peptide conjugates will make their way into the clinic within this decade.
| Material | Examples | Properties | Nanoscale properties | Ref. |
|---|---|---|---|---|
| Noble metals | Gold, silver and platinum | Biocompatible, bacteriostatic, unique electromagnetic, optical and catalytic properties | Plasmonics and hyperthermic (heating) | [65,67,116–118] |
| Semiconductor quantum dots | CdSe/ZnS core-shell quantam dots (CdTe, CdS and InP) | Ease of bioconjugation, optical properties ideal for biological sensing and imaging | Size-tunable fluorescence emission from UV to near infrared; sensitivity to charge and excellent photosensitizers | [35,36,69–70,85,89,110,115,122–123] |
| Oxides | Iron oxide | Magnetic properties for MRI | Magnetic contrast properties | [71–72,75,83,91–93,98] |
| Polymers | Poly(lactic-co-glycolic acid), poly(amido amine) and dendrimers | Biocompatibility, biodegradability and controlled release | High drug-loading capacity and controllable cargo release | [73,91,98,125] |
| Vesicles | Micelles and liposomes | Biocompatibility, biodegradability and controlled release | High drug-loading capacity (large S/V) and controllable cargo release | [5–8,72,119–120] |
| Proteins | Therapeutic proteins (e.g., Abraxane® – albumin carrier of paclitaxel) | Biocompatibility, tunable pharmacokinetics and targeting | Capable of genetic manipulation and ability to be coupled to drugs | [112,113] |
| Nucleic acids | Small interfering RNA, antisense RNA and plasmid DNA | Biocompatibility, tunable pharmacokinetics and targeting | Structural recognition (e.g., aptamers) and ability to be coupled to drugs | [73–74,99,100,103,119,120,124] |
| Peptide | Nanoparticle (size [nm]) | Targeted cells (aim of study) | Ref. |
|---|---|---|---|
| TAT | Quantum dots (4–20) | Human embryonic kidney (293), African monkey kidney (COS-1), HeLa and murine mesenchymal stem cells (intracellular labeling, in vivo fate-tracking and in vivo tumor imaging) | [35,36,69,70,134] |
| Gold particles (5–30) | Human and murine fibroblasts (intracellular labeling) | [65,67] | |
| Iron oxide NPs (45) | Human CD34+ hematopoietic cells, mouse neural progenitor cells, (C17.2), human CD4+ lymphocytes and mouse splenocytes (intracellular labeling and in vivo fate-tracking) | [71] | |
| Iron oxide NPs/PEI/ plasmid DNA (>300) | Human NT2 neural stem cells, in vivo rat spinal cord (transgene expression and in vivo magnetofection/gene expression) | [74] | |
| pH-sensitive micelles (35–50) | MCF-7 human breast cancer cells (drug delivery) | [7,8] | |
| pH-sensitive liposomes (110–160) | U-87 MG astrocytoma cells (drug delivery) | [5,6] | |
| Iron-loaded liposomes (>100) | In vivo rat model of spinal cord injury (in vivo spinal injury targeting) | [72] | |
| Bacterial magnetic NPs/PAMAM/siRNA (>100) | U251-MG glioblastoma cells (in vivo EGFR expression silencing) | [73] | |
| RGD | Quantum dots (10–15) | Osteoblast cells, MCF-7 and U87-MG (intracellular labeling and in vivo tumor imaging) | [80,85,89] |
| Iron oxide NPs (15) | Endothelial cells (intracellular labeling and in vivo tumor imaging) | [83,91–93,98] | |
| Liposomes (Gd-loaded) (>50) | Mouse tumor vasculature (in vivo tumor imaging) | [92] | |
| 125I-labeled dendrimer (15) | In vivo angiogenesis imaging (mouse) | [91] | |
| PLGA NPs (300–430) | Mouse tumor vasculature (in vivo drug delivery) | [98] | |
| Plasmid DNA (>300) | Neuronal cells (SH-SY5Y), endothelial cells (intracellular labeling) | [99,100] | |
| siRNA-PEI-PEG (∼100) | Mouse xenografts (in vivo gene silencing) | [103] | |
| Pep-1/Chariot™ | Various proteins (GFP, B-Gal, antibodies) (6–20) | Fibroblasts (HS-68, NIH 3T3, 293, Jurkat T, COS-7; intracellular labeling) | [104] |
| Quantum dots (5–10) | Fibroblasts (CHO cells; for high-throughput cell-based drug screening) | [110] | |
| Therapeutic proteins (HSP-27, SOD-1) (8–20) | Astrocytes, primary neuronal cells, in vivo gerbil model of ischemia and in vivo rat model of spinal cord injury (protection against oxidative stress) | [112,113] | |
| Organelle-specific | Quantum dots (20–50) | Vero cells (subcellular compartment labeling) | [115] |
| Au-NPs, fluorescent Au-NPs (3–20) | Osteocarcoma, HeLa, HepG2 and NIH 3T3 (intracellular labeling) | [116–118,135] | |
| Liposomes-plasmid DNA (100) | Epithelial cells (in vitro gene expression) | [119,120] | |
| Neuropeptide (Ast 1) | Quantum dots (15) | Fibroblasts (NIH 3T3 and A431; intracellular labeling) | [122,123] |
| Rabies virus-derived peptide | siRNA (5) | Neuronal cells (Neuro 2a; in vivo gene silencing) | [124] |
| PAMAM-PEG/plasmid DNA (>300) | Brain capillary endothelial cells (in vivo gene expression) | [125] |
Executive summary
Therapeutic applications of nanoparticles
▪ The small size of nanoparticles (NPs) affords them entry to areas not otherwise accessible to other materials and their large surface area allows them to carry large ‘payloads’.
▪ Peptide-mediated NP delivery is one of the most facile and noninvasive means of cellular NP delivery.
Bioconjugation of peptides to nanoparticles
▪ Attachment of peptides to NP surface homogenously with control over peptide orientation, distance and valence is key; future research will invest heavily in this area.
Peptides in therapeutic delivery of nanoparticles
▪ Naturally occurring and synthetic peptides have been demonstrated to deliver a wide array of NP materials to cells with high efficiency.
▪ Future efforts will develop ‘multifunctional’ NP–peptide constructs comprised of multiple peptide species performing separate functions or a single peptide species capable of multiple functions.
Toxicity of peptide-delivered nanoparticles
▪ Bioconjugate toxicity is dependent on the NP, peptide and the NP–peptide ensemble.
▪ Initial NP–peptide toxicity assessments have been carried out in vitro; further characterization of NP properties and standardization of NP toxicity assessments are required.
▪ Future work will focus heavily on in vivo toxicity testing in animal models.
Financial & competing interests disclosure
The authors acknowledge the CB Directorate/Physical S&T Division (ARO/DTRA), ONR, NRL and the NRL-NSI for financial support. K Boeneman acknowledges an ASEE fellowship and CE Bradburne and K Robertson acknowledge NRC fellowships through the NRL. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
Papers of special note have been highlighted as: ▪ of interest ▪▪ of considerable interest
Bibliography
- 1 Yang W, Peters JI, Williams III RO. Inhaled nanoparticles – a current review. Int. J. Pharm.356(1–2),239–247 (2008).
- 2 Ganta S, Devalapally H, Shahiwala A, Amiji M. A review of stimuli-responsive nanocarriers for drug and gene delivery. J. Control. Release126(3),187–204 (2008).▪ Review of stimuli-responsive nanoparticle carriers for drug and gene delivery.
- 3 Jabr-Milane L, van Vlerken L, Devalapally H et al. Multi-functional nanocarriers for targeted delivery of drugs and genes. J. Control. Release130(2),121–128 (2008).
- 4 Zhou SF, Di YM, Chan E et al. Clinical pharmacogenetics and potential application in personalized medicine. Curr. Drug Metab.9(8),738–784 (2008).
- 5 Sawant RM, Hurley JP, Salmaso S et al. ‘SMART’ drug delivery systems: double-targeted pH-responsive pharmaceutical nanocarriers. Bioconjugate Chem.17(4),943–949 (2006).
- 6 Kale AA, Torchilin VP. Design, synthesis, and characterization of pH-sensitive PEG–PE conjugates for stimuli-sensitive pharmaceutical nanocarriers: the effect of substitutes at the hydrazone linkage on the pH stability of PEG–PE conjugates. Bioconjugate Chem.18(2),363–370 (2007).
- 7 Sethuraman VA, Bae YH. Tat peptide-based micelle system for potential active targeting of anti-cancer agents to acidic solid tumors. J. Control. Release118(2),216–224 (2007).
- 8 Sethuraman VA, Lee MC, Bae YH. A biodegradable pH-sensitive micelle system for targeting acidic solid tumors. Pharm. Res.25(3),657–666 (2008).
- 9 Liu K-K, Wang C-C, Cheng C-L, Chao J-I. Endocytic carboxylated nanodiamond for the labeling and tracking of cell division and differentiation in cancer and stem cells. Biomaterials30(26),4249–4259 (2009).
- 10 Jaiswal JK, Mattoussi H, Mauro JM, Simon SM. Long-term multiple color imaging of live cells using quantum dot bioconjugates. Nat. Biotechnol.21(1),47–51 (2003).
- 11 Shukla R, Bansal V, Chaudhary M, Basu A, Bhonde RR, Sastry M. Biocompatibility of gold nanoparticles and their endocytotic fate inside the cellular compartment: a microscopic overview. Langmuir21(23),10644–10654 (2005).
- 12 Chen FQ, Gerion D. Fluorescent CdSe/ZnS nanocrystal-peptide conjugates for long-term, nontoxic imaging and nuclear targeting in living cells. Nano Lett.4(10),1827–1832 (2004).
- 13 Lin J, Chen R, Feng S et al. Rapid delivery of silver nanoparticles into living cells by electroporation for surface-enhanced Raman spectroscopy. Biosens. Bioelectron.25(2),388–394 (2009).
- 14 Kanazawa T, Takashima Y, Murakoshi M, Nakai Y, Okada H. Enhancement of gene transfection into human dendritic cells using cationic PLGA nanospheres with a synthesized nuclear localization signal. Int. J. Pharm.379(1),187–195 (2009).
- 15 Atul P, Soma P, Kailash Chand G. Recent trends in non-viral vector-mediated gene delivery. Biotechnol. J.4(11),1559–1572 (2009).
- 16 Zhang Y, Yu LC. Single-cell microinjection technology in cell biology. Bioessays30(6),606–610 (2008).
- 17 Zhang Y, Yu LC. Microinjection as a tool of mechanical delivery. Curr. Opin. Biotech.19(5),506–510 (2008).
- 18 Yao N, Xiao W, Wang X et al. Discovery of targeting ligands for breast cancer cells using the one-bead one-compound combinatorial method. J. Med. Chem.52(1),126–133 (2009).
- 19 Medintz I. Universal tools for biomolecular attachment to surfaces. Nat. Mater.5(11),842–842 (2006).
- 20 Delehanty JB, Mattoussi H, Medintz IL. Delivering quantum dots into cells: strategies, progress and remaining issues. Anal. Bioanal. Chem.393(4),1091–1105 (2009).▪ Critical current review of available methods for quantum dot delivery to cells.
- 21 Delehanty JB, Boeneman K, Bradburne CE, Robertson K, Medintz IL. Quantum dots: a powerful tool for understanding the intricacies of nanoparticle-mediated drug delivery. Expert Opin. Drug Del.6(10),1091–1112 (2009).▪▪ Comprehensive survey of the available literature on the use of quantum dots as representative nanoparticles for nanoparticle-mediated drug design.
- 22 Aubin-Tam ME, Hamad-Schifferli K. Structure and function of nanoparticle-protein conjugates. Biomed. Mater.3(3),034001 (2008).▪▪ Comprehensive primer on the important aspects to be taken into consideration when functionalizing nanoparticles with proteins.
- 23 Bioconjugate Techniques. Hermanson GT (Ed.). Academic Press, San Diego, CA, USA (2008).
- 24 Sapsford KE, Berti L, Medintz IL. Fluorescence spectroscopy: applications in chemical biology. In: Wiley Encyclopedia of Chemical Biology. Begley TP (Ed.). Wiley Publishers, NY, USA (2008).
- 25 Rozenzhak SM, Kadakia MP, Caserta TM, Westbrook TR, Stone MO, Naik RR. Cellular internalization and targeting of semiconductor quantum dots. Chem. Comm. (17),2217–2219 (2005).
- 26 Wagner SC, Roskamp M, Colfen H, Bottcher C, Schlecht S, Koksch B. Switchable electrostatic interactions between gold nanoparticles and coiled coil peptides direct colloid assembly. Org. Biomol. Chem.7(1),46–51 (2009).
- 27 Daniel MC, Astruc D. Gold nanoparticles: assembly, supramolecular chemistry, quantum-size-related properties, and applications toward biology, catalysis, and nanotechnology. Chem. Rev.104(1),293–346 (2004).
- 28 Love JC, Estroff LA, Kriebel JK, Nuzzo RG, Whitesides GM. Self-assembled monolayers of thiolates on metals as a form of nanotechnology. Chem. Rev.105(4),1103–1169 (2005).
- 29 Pinaud F, King D, Moore H-P, Weiss S. Bioactivation and cell targeting of semiconductor CdSe/ZnS nanocrystals with phytochelatin-related peptides. J. Am. Chem. Soc.126(19),6115–6123 (2004).
- 30 Dif A, Boulmedais F, Pinot M et al. Small and stable peptidic PEGylated quantum dots to target polyhistidine-tagged proteins with controlled stoichiometry. J. Am. Chem. Soc.131(41),14738–14746 (2009).
- 31 Hochuli E, Bannwarth W, Dobeli H, Gentz R, Stuber D. Genetic approach to facilitate purification of recombinant proteins with a novel metal chelate adsorbent. Biotechnol.6,1321–1325 (1988).
- 32 Sapsford KE, Pons T, Medintz IL et al. Kinetics of metal-affinity driven self-assembly between proteins or peptides and CdSe-ZnS quantum dots. J. Phys. Chem. C, 111(31),11528–11538 (2007).
- 33 Medintz IL, Clapp AR, Brunel FM et al. Proteolytic activity monitored by fluorescence resonance energy transfer through quantum-dot-peptide conjugates. Nat. Mater.5(7),581–589 (2006).
- 34 Sapsford KE, Farrell D, Sun S, Rasooly A, Mattoussi H, Medintz IL. Monitoring of enzymatic proteolysis on a electroluminescent-CCD microchip platform using quantum dot-peptide substrates. Sensor. Actuat. B-Chem.139(1),13–21 (2009).
- 35 Delehanty JB, Medintz IL, Pons T, Brunel FM, Dawson PE, Mattoussi H. Self-assembled quantum dot-peptide bioconjugates for selective intracellular delivery. Bioconjugate Chem.17(4),920–927 (2006).
- 36 Medintz IL, Pons T, Delehanty JB et al. Intracellular delivery of quantum dot-protein cargos mediated by cell penetrating peptides. Bioconjugate Chem.19(9),1785–1795 (2008).
- 37 Xu C, Xu K, Gu H et al. Dopamine as a robust anchor to immobilize functional molecules on the iron oxide shell of magnetic nanoparticles. J. Am. Chem. Soc.126(32),9938–9939 (2004).
- 38 Sarikaya M, Tamerler C, Schwartz DT, Baneyx FO. Materials assembly and formation using engineered polypeptides. Ann. Rev. Mater. Res.34,373–408 (2004).
- 39 Peelle BR, Krauland EM, Wittrup KD, Belcher AM. Design criteria for engineering inorganic material-specific peptides. Langmuir21(15),6929–6933 (2005).
- 40 Hainfeld JF, Liu W, Halsey C, Freimuth P, Powell RD. Ni-NTA-gold clusters target His-tagged proteins. J. Struct. Biol.127(2),185–198 (1999).
- 41 Kim J, Park HY, Kim J et al. Ni-nitrilotriacetic acid-modified quantum dots as a site-specific labeling agent of histidine-tagged proteins in live cells. Chem. Comm. (16),1910–1912 (2008).
- 42 Patel JD, O’Carra R, Jones J, Woodward JG, Mumper RJ. Preparation and characterization of nickel nanoparticles for binding to his-tag proteins and antigens. Pharmaceut. Res.24(2),343–352 (2007).
- 43 Kim SH, Jeyakumar M, Katzenellenbogen JA. Dual-mode fluorophore-doped nickel nitrilotriacetic acid-modified silica nanoparticles combine histidine-tagged protein purification with site-specific fluorophore labeling. J. Am. Chem. Soc.129(43),13254–13264 (2007).
- 44 Medintz IL, Berti L, Pons T et al. A reactive peptidic linker for self-assembling hybrid quantum dot-DNA bioconjugates. Nano Lett.7(6),1741–1748 (2007).
- 45 Wolfgang HB, Robert S. ‘Click’ chemistry in polymer and material science: an update. Macromo. Rapid Comm.29(12–13),952–981 (2008).
- 46 Prasuhn DE, Blanco-Canosa JB, Vora GJ et al. Combining chemoselective ligation with polyhistidine-driven self-assembly for the modular display of biomolecules on quantum dots. ACS Nano4(1),267–278 (2010).
- 47 Chen I, Ting AY. Site-specific labeling of proteins with small molecules in live cells. Curr. Opin. Biotech.16(1),35–40 (2005).
- 48 Stachler MD, Chen I, Ting AY, Bartlett JS. Site-specific modification of AAV vector particles with biophysical probes and targeting ligands using biotin ligase. Mol. Ther.16(8),1467–1473 (2008).
- 49 Pons T, Medintz IL, Sapsford KE et al. On the quenching of semiconductor quantum dot photoluminescence by proximal gold nanoparticles. Nano Lett.7(10),3157–3164 (2007).
- 50 Green M, Loewenstein PM. Autonomous functional domains of chemically synthesized human immunodeficiency virus Tat trans-activator protein. Cell55(6),1179–1188 (1988).
- 51 Frankel AD, Pabo CO. Cellular uptake of the Tat protein from human immunodeficiency virus. Cell55(6),1189–1193 (1988).
- 52 Vives E, Brodin P, Lebleu B. A truncated HIV-1 Tat protein basic domain rapidly translocates through the plasma membrane and accumulates in the cell nucleus. J. Biol. Chem.272(25),16010–16017 (1997).
- 53 Schwarze SR, Hruska KA, Dowdy SF. Protein transduction: unrestricted delivery into all cells? Trends Cell Biol.10(7),290–295 (2000).
- 54 Vives E. Cellular uptake of the Tat peptide: an endocytosis mechanism following ionic interactions. J. Mol. Recognit.16(5),265–271 (2003).
- 55 Vives E, Richard JP, Rispal C, Lebleu B. Tat peptide internalization: seeking the mechanism of entry. Curr. Protein Pept. Sci.4(2),125–132 (2003).
- 56 Vivès E, Schmidt J, Pèlegrin A. Cell-penetrating and cell-targeting peptides in drug delivery. BBA Rev. Cancer, 1786(2),126–138 (2008).▪▪ Review of cell-penetrating peptides for use in drug delivery applications.
- 57 Joliot A, Pernelle C, Deagostini-Bazin H, Prochiantz A. Antennapedia homeobox peptide regulates neural morphogenesis. Proc. Natl Acad. Sci. USA88(5),1864–1868 (1991).
- 58 Elliott G, O’Hare P. Intercellular trafficking and protein delivery by a herpesvirus structural protein. Cell88(2),223–233 (1997).
- 59 Oehlke J, Scheller A, Wiesner B et al. Cellular uptake of an α-helical amphipathic model peptide with the potential to deliver polar compounds into the cell interior non-endocytically. Biochim. Biophys. Acta1414(1–2),127–139 (1998).
- 60 Futaki S, Suzuki T, Ohashi W et al. Arginine-rich peptides. an abundant source of membrane-permeable peptides having potential as carriers for intracellular protein delivery. J. Biol. Chem.276(8),5836–5840 (2001).
- 61 Console S, Marty C, Garcia-Echeverria C, Schwendener R, Ballmer-Hofer K. Antennapedia and HIV transactivator of transcription (TAT) ‘protein transduction domains’ promote endocytosis of high molecular weight cargo upon binding to cell surface glycosaminoglycans. J. Biol. Chem.278(37),35109–35114 (2003).
- 62 Lundberg M, Wikstrom S, Johansson M. Cell surface adherence and endocytosis of protein transduction domains. Mol. Ther.8(1),143–150 (2003).
- 63 Gupta B, Levchenko TS, Torchilin VP. Intracellular delivery of large molecules and small particles by cell-penetrating proteins and peptides. Adv. Drug. Deliv. Rev.57(4),637–651 (2005).
- 64 Tyagi M, Rusnati M, Presta M, Giacca M. Internalization of HIV-1 Tat requires cell surface heparan sulfate proteoglycans. J. Biol. Chem.276(5),3254–3261 (2001).
- 65 Tkachenko AG, Xie H, Liu Y et al. Cellular trajectories of peptide-modified gold particle complexes: comparison of nuclear localization signals and peptide transduction domains. Bioconjugate Chem.15(3),482–490 (2004).
- 66 de la Fuente JM, Fandel M, Berry CC et al. Quantum dots protected with tiopronin: a new fluorescence system for cell-biology studies. Chembiochem.6(6),989–991 (2005).
- 67 Berry CC, de la Fuente JM, Mullin M, Chu SWL, Curtis ASG. Nuclear localization of HIV-1 tat functionalized gold nanoparticles. IEEE T. Nanobiosci.6(4),262–269 (2007).
- 68 Richard JP, Melikov K, Vives E et al. Cell-penetrating peptides. A re-evaluation of the mechanism of cellular uptake. J. Biol. Chem.278(1),585–590 (2003).
- 69 Ruan G, Agrawal A, Marcus AI, Nie S. Imaging and tracking of tat peptide-conjugated quantum dots in living cells: new insights into nanoparticle uptake, intracellular transport, and vesicle shedding. J. Am. Chem. Soc.129(47),14759–14766 (2007).
- 70 Lei Y, Tang H, Yao L, Yu R, Feng M, Zou B. Applications of mesenchymal stem cells labeled with Tat peptide conjugated quantum dots to cell tracking in mouse body. Bioconjugate Chem.19(2),421–427 (2008).
- 71 Lewin M, Carlesso N, Tung CH et al. Tat peptide-derivatized magnetic nanoparticles allow in vivo tracking and recovery of progenitor cells. Nat. Biotechnol.18(4),410–414 (2000).
- 72 Liu Y, Wang CY, Kong XH et al. Novel multifunctional polyethylene glycol-transactivating-transduction protein-modified liposomes cross the blood-spinal cord barrier after spinal cord injury. J. Drug Target.18(6),420–429 (2009).
- 73 Han L, Zhang A, Wang H, Pu P, Kang C, Chang J. Tat-BMPs-PAMAM conjugates enhance therapeutic effect of small interference RNA on U251 glioma cells in vitro and in vivo. Hum. Gene Ther.21(4),417–426 (2009).
- 74 Song HP, Yang JY, Lo SL et al. Gene transfer using self-assembled ternary complexes of cationic magnetic nanoparticles, plasmid DNA and cell-penetrating Tat peptide. Biomaterials31(4),769–778 (2009).
- 75 Engel J, Odermatt E, Engel A et al. Shapes, domain organizations and flexibility of laminin and fibronectin, two multifunctional proteins of the extracellular matrix. J. Mol. Biol.150(1),97–120 (1981).
- 76 Hood JD, Cheresh DA. Role of integrins in cell invasion and migration. Nat. Rev. Cancer, 2(2),91–100 (2002).
- 77 Desgrosellier JS, Cheresh DA. Integrins in cancer: biological implications and therapeutic opportunities. Nat. Rev. Cancer, 10(1),9–22 (2010).
- 78 Di Minno G, Coppola A, Di Minno MN, Poon MC. Glanzmann’s thrombasthenia (defective platelet integrin αIIbβ3): proposals for management between evidence and open issues. Thromb. Haemostasis, 102(6),1157–1164 (2009).
- 79 Mizejewski GJ. Role of integrins in cancer: survey of expression patterns. P. Soc. Exp. Biol. Med.222(2),124–138 (1999).
- 80 Lieleg O, Lopez-Garcia M, Semmrich C, Auernheimer J, Kessler H, Bausch AR. Specific integrin labeling in living cells using functionalized nanocrystals. Small3(9),1560–1565 (2007).
- 81 Ko MH, Kim S, Kang WJ et al. In vitro derby imaging of cancer biomarkers using quantum dots. Small5(10),1207–1212 (2009).
- 82 Zako T, Nagata H, Terada N et al. Cyclic RGD peptide-labeled upconversion nanophosphors for tumor cell-targeted imaging. Biochem. Biophys. Res. Co.381(1),54–58 (2009).
- 83 Montet X, Montet-Abou K, Reynolds F, Weissleder R, Josephson L. Nanoparticle imaging of integrins on tumor cells. Neoplasia8(3),214–222 (2006).
- 84 Chen K, Xie J, Xu H et al. Triblock copolymer coated iron oxide nanoparticle conjugate for tumor integrin targeting. Biomaterials30(36),6912–6919 (2009).
- 85 Cai W, Shin DW, Chen K et al. Peptide-labeled near-infrared quantum dots for imaging tumor vasculature in living subjects. Nano Lett.6(4),669–676 (2006).
- 86 Shah BS, Clark PA, Moioli EK, Stroscio MA, Mao JJ. Labeling of mesenchymal stem cells by bioconjugated quantum dots. Nano Lett.7(10),3071–3079 (2007).
- 87 Srinivasan R, Marchant RE, Gupta AS. In vitro and in vivo platelet targeting by cyclic RGD-modified liposomes. J. Biomed. Mater. Res.93(3),1004–1015 (2009) (Epub ahead of print).
- 88 Temming K, Schiffelers RM, Molema G, Kok RJ. RGD-based strategies for selective delivery of therapeutics and imaging agents to the tumour vasculature. Drug Resist. Update8(6),381–402 (2005).
- 89 Smith BR, Cheng Z, De A, Koh AL, Sinclair R, Gambhir SS. Real-time intravital imaging of RGD-quantum dot binding to luminal endothelium in mouse tumor neovasculature. Nano Lett.8(9),2599–2606 (2008).
- 90 Cai W, Chen X. Preparation of peptide-conjugated quantum dots for tumor vasculature-targeted imaging. Nat. Protoc.3(1),89–96 (2008).
- 91 Almutairi A, Rossin R, Shokeen M et al. Biodegradable dendritic positron-emitting nanoprobes for the noninvasive imaging of angiogenesis. Proc. Natl Acad. Sci. USA106(3),685–690 (2009).
- 92 van Tilborg GA, Mulder WJ, van der Schaft DW et al. Improved magnetic resonance molecular imaging of tumor angiogenesis by avidin-induced clearance of nonbound bimodal liposomes. Neoplasia10(12),1459–1469 (2008).
- 93 Schottelius M, Laufer B, Kessler H, Wester HJ. Ligands for mapping αvβ3 integrin expression in vivo. Accounts Chem. Res.42(7),969–980 (2009).
- 94 Montet X, Funovics M, Montet-Abou K, Weissleder R, Josephson L. Multivalent effects of RGD peptides obtained by nanoparticle display. J. Med. Chem.49(20),6087–6093 (2006).
- 95 Murphy EA, Majeti BK, Barnes LA et al. Nanoparticle-mediated drug delivery to tumor vasculature suppresses metastasis. Proc. Natl Acad. Sci. USA105(27),9343–9348 (2008).
- 96 Wang Z, Chui WK, Ho PC. Design of a multifunctional PLGA nanoparticulate drug delivery system: evaluation of its physicochemical properties and anticancer activity to malignant cancer cells. Pharm. Res.26(5),1162–1171 (2009).
- 97 Danhier F, Vroman B, Lecouturier N et al. Targeting of tumor endothelium by RGD-grafted PLGA-nanoparticles loaded with paclitaxel. J. Control. Release140(2),166–173 (2009).
- 98 Sugahara KN, Teesalu T, Karmali PP et al. Tissue-penetrating delivery of compounds and nanoparticles into tumors. Cancer Cell16(6),510–520 (2009).
- 99 Suk JS, Suh J, Choy K, Lai SK, Fu J, Hanes J. Gene delivery to differentiated neurotypic cells with RGD and HIV Tat peptide functionalized polymeric nanoparticles. Biomaterials27(29),5143–5150 (2006).
- 100 Zuber G, Dontenwill M, Behr JP. Synthetic virus-like particles for targeted gene delivery to αvβ3 integrin-presenting endothelial cells. Mol. Pharmacol.6(5),1544–1552 (2009).
- 101 Zhou QH, Wu C, Manickam DS, Oupicky D. Evaluation of pharmacokinetics of bioreducible gene delivery vectors by real-time PCR. Pharmceut. Res.26(7),1581–1589 (2009).
- 102 Wang XL, Xu R, Wu X, Gillespie D, Jensen R, Lu ZR. Targeted systemic delivery of a therapeutic siRNA with a multifunctional carrier controls tumor proliferation in mice. Mol. Pharmacol.6(3),738–746 (2009).
- 103 Schiffelers RM, Ansari A, Xu J et al. Cancer siRNA therapy by tumor selective delivery with ligand-targeted sterically stabilized nanoparticle. Nucleic Acids Res.32(19),e149 (2004).
- 104 Morris MC, Depollier J, Mery J, Heitz F, Divita G. A peptide carrier for the delivery of biologically active proteins into mammalian cells. Nat. Biotechnol.19(12),1173–1176 (2001).
- 105 Simeoni F, Morris MC, Heitz F, Divita G. Insight into the mechanism of the peptide-based gene delivery system MPG: implications for delivery of siRNA into mammalian cells. Nucleic Acids Res.31(11),2717–2724 (2003).
- 106 Morris MC, Gros E, Aldrian-Herrada G et al. A non-covalent peptide-based carrier for in vivo delivery of DNA mimics. Nucleic Acids Res.35(7),e49 (2007).
- 107 Deshayes S, Heitz A, Morris MC, Charnet P, Divita G, Heitz F. Insight into the mechanism of internalization of the cell-penetrating carrier peptide Pep-1 through conformational analysis. Biochemistry43(6),1449–1457 (2004).▪ Provides mechanistic insight into the mechanism of Pep-1 peptide internalization by cells.
- 108 Munoz-Morris MA, Heitz F, Divita G, Morris MC. The peptide carrier Pep-1 forms biologically efficient nanoparticle complexes. Biochem. Biophys. Res. Co.355(4),877–882 (2007).
- 109 Morris MC, Deshayes S, Heitz F, Divita G. Cell-penetrating peptides: from molecular mechanisms to therapeutics. Biol. Cell100(4),201–217 (2008).
- 110 Mattheakis LC, Dias JM, Choi YJ et al. Optical coding of mammalian cells using semiconductor quantum dots. Anal. Biochem.327(2),200–208 (2004).
- 111 Chang JC, Su HL, Hsu SH. The use of peptide-delivery to protect human adipose-derived adult stem cells from damage caused by the internalization of quantum dots. Biomaterials29(7),925–936 (2008).
- 112 An JJ, Lee YP, Kim SY et al. Transduced human PEP-1-heat shock protein 27 efficiently protects against brain ischemic insult. FEBS J.275(6),1296–1308 (2008).
- 113 Yune TY, Lee JY, Jiang MH, Kim DW, Choi SY, Oh TH. Systemic administration of PEP-1-SOD1 fusion protein improves functional recovery by inhibition of neuronal cell death after spinal cord injury. Free Radical Biol. Med.45(8),1190–1200 (2008).
- 114 Zhang YE, Wang JN, Tang JM et al. In vivo protein transduction: delivery of PEP-1-SOD1 fusion protein into myocardium efficiently protects against ischemic insult. Mol. Cell.27(2),159–166 (2009).
- 115 Hoshino A, Fujioka K, Oku T et al. Quantum dots targeted to the assigned organelle in living cells. Microbiol. Immunol.48(12),985–994 (2004).
- 116 Lin SY, Chen NT, Sum SP, Lo LW, Yang CS. Ligand exchanged photoluminescent gold quantum dots functionalized with leading peptides for nuclear targeting and intracellular imaging. Chem. Comm.39,4762–4764 (2008).
- 117 Xie W, Wang L, Zhang Y et al. Nuclear targeted nanoprobe for single living cell detection by surface-enhanced Raman scattering. Bioconjugate Chem.20(4),768–773 (2009).
- 118 Mandal D, Maran A, Yaszemski MJ, Bolander ME, Sarkar G. Cellular uptake of gold nanoparticles directly cross-linked with carrier peptides by osteosarcoma cells. J. Mat. Sci.20(1),347–350 (2009).
- 119 Boulanger C, Di Giorgio C, Vierling P. Synthesis of acridine-nuclear localization signal (NLS) conjugates and evaluation of their impact on lipoplex and polyplex-based transfection. Eur. J. Med. Chem.40(12),1295–1306 (2005).
- 120 Wiseman JW, Scott ES, Shaw PA, Colledge WH. Enhancement of gene delivery to human airway epithelial cells in vitro using a peptide from the polyoma virus protein VP1. J. Gene Med.7(6),759–770 (2005).
- 121 Lenz C, Sondergaard L, Grimmelikhuijzen CJ. Molecular cloning and genomic organization of a novel receptor from Drosophila melanogaster structurally related to mammalian galanin receptors. Biochem. Biophys. Res. Co.269(1),91–96 (2000).
- 122 Biju V, Muraleedharan D, Nakayama K et al. Quantum dot-insect neuropeptide conjugates for fluorescence imaging, transfection, and nucleus targeting of living cells. Langmuir23(20),10254–10261 (2007).
- 123 Anas A, Okuda T, Kawashima N et al. Clathrin-mediated endocytosis of quantum dot-peptide conjugates in living cells. ACS Nano3(8),2419–2429 (2009).
- 124 Kumar P, Wu H, McBride JL et al. Transvascular delivery of small interfering RNA to the central nervous system. Nature448(7149),39–43 (2007).
- 125 Liu Y, Huang R, Han L et al. Brain-targeting gene delivery and cellular internalization mechanisms for modified rabies virus glycoprotein RVG29 nanoparticles. Biomaterials30(25),4195–4202 (2009).
- 126 Chang E, Thekkek N, Yu WW, Colvin VL, Drezek R. Evaluation of quantum dot cytotoxicity based on intracellular uptake. Small2(12),1412–1417 (2006).
- 127 Warheit DB. How meaningful are the results of nanotoxicity studies in the absence of adequate material characterization? Toxicol. Sci.101(2),183–185 (2008).
- 128 Hood E. Nanotechnology: looking as we leap. Environ Health Perspect, 112(13),A740–749 (2004).
- 129 Lewinski N, Colvin V, Drezek R. Cytotoxicity of nanoparticles. Small4(1),26–49 (2008).▪▪ Survey of the available literature on the cytotoxicity of multiple classes of nanoparticles.
- 130 Duconge F, Pons T, Pestourie C et al. Fluorine-18-labeled phospholipid quantum dot micelles for in vivo multimodal imaging from whole body to cellular scales. Bioconjugate Chem.19(9),1921–1926 (2008).
- 131 Duan H, Nie S. Cell-penetrating quantum dots based on multivalent and endosome-disrupting surface coatings. J. Am. Chem. Soc.129(11),3333–3338 (2007).
- 132 Kato T, Okada S, Yutaka T, Yabuuchi H. The effects of sucrose loading on lysosomal hydrolases. Mol. Cell. Biochem.60(1),83–98 (1984).
- 133 Ferrari V, Cutler DJ. Kinetics and thermodynamics of chloroquine and hydroxy-chloroquine transport across the human erythrocyte membrane. Biochem. Pharmacol.41(1),23–30 (1991).
- 134 Stroh M, Zimmer JP, Duda DG et al. Quantum dots spectrally distinguish multiple species within the tumor milieu in vivo. Nat. Med.11(6),678–682 (2005).
- 135 Tkachenko AG, Xie H, Liu Y et al. Cellular trajectories of peptide-modified gold particle complexes: comparison of nuclear localization signals and peptide transduction domains. Bioconj. Chem.15(3),482–490 (2004).
- 201 Invitrogen Corp. Molecular Probes: the handbook (2010) www.invitrogen.com/site/us/en/home/References/Molecular-Probes-The-Handbook.html


