








Facile approach for constructing TEV insertions to probe protein structure in vivo
Abstract
The tobacco etch virus (TEV) protease has been used as a tool to examine protein structure in vivo. TEV cleavage sites (TEVcs) have been introduced via cloning into unique restriction sites or random transposon mutagenesis. We describe a facile, efficient method for introducing TEVcs at precise locations in a gene to test specific predictions about protein structure. The method uses the λ Red recombination system to construct seamless, in-frame insertions of the TEVcs at any desired location within an open reading frame (ORF). The system was tested using the multifunctional PutA protein Salmonella enterica sv. Typhimurium. The first step involved insertion of a chloramphenicol resistance (CamR) cassette with a transcriptional terminator at the desired location. A second swap then replaces the CamR insertion with the TEVcs. Placing a copy of the lac operon downstream of the putA gene provides a simple counterselection for replacement of the CamR insertion and also provides a reporter gene for monitoring transcription of the mutated gene.
Introduction
The tobacco etch virus (TEV) protease provides an in vivo tool to study protein topology, by identifying exposed versus buried residues or intracellular versus extracellular domains of membrane proteins. The TEV protease recognizes a rare seven amino acid sequence glu-asp-leutyr-phe-gln-ser and cleaves between the gln-ser residues (1). Previous research suggested that insertion of the recognition site does not interfere with protein function due to its small size (2). This led to the exploitation of TEV protease as a tool for dissecting functional domains of proteins.
Ehrmann et al. (2) designed an elegant transposon-based approach for insertion of TEV cleavage sites (TEVcs) into multiple sites in a gene. With a large number of insertions, this approach can provide useful information on the topology of certain proteins in vivo. However, it does not allow directed insertions of the TEVcs at defined sites to test specific predictions about protein topology. We developed an alternative method for rapidly constructing insertions at defined sites in an open reading frame (ORF). This approach is based upon three genetic tricks: (i) use of the λ Red recombination system to construct directed insertions of a selectable marker; (ii) use of the λ Red recombination system to replace the selectable marker with a short, nonselectable sequence; and (iii) use of an in-frame, downstream reporter to enrich for replacements of the desired type.
The λ Red recombination system provides an efficient genetic tool for introducing TEVcs insertions at specific sites in a gene. Although Red-mediated genetic exchange is specific and efficient, strong counterselections for insertion of seamless mutations are limited. When used for such singlecopy substitutions, commonly used counterselectable markers yield false positive results so frequently that isolation of desired mutants demands labor-intensive secondary screening. In contrast, placement of an in-frame copy of the lac operon under transcriptional control of the target gene provides a clean and robust counterselection (Figure 1). The lac insertion also provides a tool for monitoring the effect of the insertions on transcription. Coupling these two genetic tricks provides an efficient approach to introduce seamless, in-frame insertions of the TEVcs into a specific location in the target gene. Once TEVcs insertions are confirmed, TEV protease cleavage assays can be done in vivo or with purified protein in vitro to determine the accessibility of TEVcs in the protein.
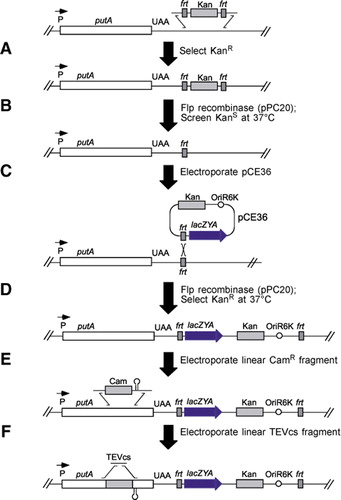
First, an frt scar was constructed downstream of putA as shown in panels A and B. Then, the lac operon was inserted between the translational and transcriptional stop sites of putA via pCE36. Insertion of pCE36 into the chromosome has several benefits. The lac operon provides a counterselectable marker for insertion of TEVcs or for constructing unmarked point mutants, and it serves as a reporter gene for assaying transcriptional regulation of the upstream gene. The kanamycin resistance (KanR) marker on the plasmid provides a direct selection for the desired insertions by simply plating on Kan 5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside (X-Gal) plates. All KanR X-Gal+ colonies selected contained pCE36 in the proper location and orientation. Because the plasmid also places KanR adjacent to the mutated gene, it also provides a selectable marker for moving the mutants into new strain backgrounds. For example, when placed downstream of putA, KanR was very tightly linked to the TEVcs insertion via P22 transduction. Following construction of the downstream lac KanR insertion, two sequential exchanges catalyzed by the λ Red system were used to insert the TEVcs into selected positions within putA as shown in panels E and F. The initial exchange involved insertion of a cassette carrying a chloramphenicol resistance (CamR) gene and a strong transcriptional terminator. These insertions were obtained by selecting for CamR and screening for X-Gal-. The terminator prevents expression of the downstream lac operon, hence the CamR insertion mutants were unable to grow on lactose as a sole carbon source. A second exchange then replaced the CamR cassette with a short DNA fragment including the TEVcs. Although these exchanges did not introduce a selectable marker, the desired replacements could be selected by demanding growth on lactose as a sole carbon source.
We tested this method on the PutA protein from Salmonella enterica sv. Typhimurium. PutA is a multifunctional protein involved in both proline degradation and autogenous regulation of the put operon. In the absence of proline, PutA dimerizes and binds DNA to repress transcription of the put operon. In the presence of proline, PutA associates with the membrane and catalyzes the oxidation of proline to glutamate. While a great deal is known about the enzymatic activities of PutA, little is known of PutA topology, and few distinct domains have yet been assigned to these activities. Recently, the crystal structure of a truncated form of the Escherichia coli PutA (PutA669) protein has been solved, including the 669 amino acids of the N terminus (3). The crystal structure of PutA669 led to predictions of domain organization. Insertion of TEVcs into PutA allowed tests of some of these predictions in vivo.
Materials and methods
Strains and Media
When required, antibiotic concentrations in rich media were as follows: 90 µg/mL ampicillin, 20 µg/mL chloramphenicol, and 50 µg/mL kanamycin (all from Sigma, St. Louis, MO, USA). In minimal media, a lower concentration of ampicillin (15 µg/mL) was used. Minimal media were prepared with sodium succinate as a carbon source supplemented with or without 0.2% proline. Lactose plates were prepared as no citrate E (NCE) media plates supplemented with 0.4% lactose (4).
Construction of a π-Dependent Vector for Use as a PCR Template
In order to produce a template for PCR amplification, the TEVcs was cloned into π-dependent plasmid pGP704 (5). The TEVcs was created by annealing oligonucleotides of the corresponding amino acid sequence of a wild-type cleavage site. Complementary oligonucleotides were designed with flanking EcoRI and XbaI sticky ends as follows: 5′-CTAGAGAAAACCTGTATTTTCAGAGCG-3′ and 5′-AATTCGCTCTGAAAATACAGGTTTTCT-3′. pGP704 was digested with XbaI and EcoRI (Fermentas, Hanover, MD, USA) according to manufacturer's recommendations. Ligation of pGP704 and the TEVcs yielded plasmid pPC265.
Creating a Chloramphenicol Cassette PCR Template
Demanding growth on lactose as a counterselection required that the upstream chloramphenicol resistance cassette inserted into putA had a strong ρ-independent transcriptional terminator collectively called CamR. CamR was created by adding the ρ- independent terminator from putA to the chloramphenicol cassette. Primers 5′-CATCTTGGTTACCGTGAAGTTA CCATCACGGAAAAAGGTTATGCT GTGTAGGCTGGAGCTGCTTCG-3′ and 5′-TGGGTTATCAAGAGGGTCA TTATATTTCGCGGAATAACATCAT TTGGTGACGAGTAAAAAGGCTC CATTGACGGA-3′ were used to PCR amplify the chloramphenicol cassette from pKD3 (6) flanked by 52 bases of homology to putA sequence between the transcriptional and translational stop site. The PCR product was then used for recombination via the λ Red system into S. Typhimurium as previously described (6). CamR clones were restreaked and stored for future use.
PCR Amplification of Cassettes for Recombination and Creation of Mutants
The method of Ellermeier et al. (7) was used to insert the lac operon between the translational and transcriptional stop sites of putA. The resulting strain was then used to construct TEVcs insertions. Appropriate primer pairs were used to amplify either CamR or the TEVcs for recombination with flanking homology to putA. Each CamR primer pair had 18 bp annealing to the sequence of CamR and a 5′ or 3′ overhang of 52 bp with homology to putA. All PCR products were separated on a 0.8% agarose gel and purified using a QIAprep® PCR Purification kit (Qiagen, Valencia, CA, USA) before use. TEVcs were inserted through two rounds of λ Red recombination. The first round was completed by inserting CamR into putA in-frame at the desired site. Chloramphenicolresistant colonies were then patched to kanamycin, ampicillin, and minimal plates with lactose as the sole carbon source. Recombinants containing the CamR in the proper place failed to grow on minimal lactose plates. A second round of λ Red-induced recombination replaced the CamR with the TEVcs by demanding growth on minimal media plates with lactose as a sole carbon source. Lac+ colonies were screened for proper insertion of the TEVcs by PCR using hybrid primers that annealed to the TEVcs and to the flanking putA for an expected product size of 120 bp. The putA::TEVcs were transduced into a clean background by selection for the tightly linked kanamycin resistance (KanR) (4). KanR colonies were rescreened for the presence of TEVcs by PCR amplification.
Cleavage and Enzyme Assays
Mutants containing the plasmid pGEX-2T (8) expressing a glutathione S-transferase (GST)-TEV protease hybrid were grown overnight and subcultured at a dilution of 1:10 into E-minimal medium with succinate and ampicillin plus proline. Cells were grown to exponential phase (A600 approximately 0.4), then isopropyl β-D-1-thiogalactopyranoside (IPTG) was added to a final concentration of 10 µM. After a 4-h induction, cells were centrifuged for 5 min at 10,000× g, and resuspended in phosphate-buffered saline (PBS). Lysozyme was added to a final concentration of 1 mg/mL, and cultures were left on ice for 30 min. Sonication and Western blot analyses were done using standard procedures as described in Reference 9. In vivo proline dehydrogenase (10) and β-galactosidase (4) assays were performed as previously described.
Western Blot Analyses
Four to twenty percent NOVEX® Tris-Glycine gels (Invitrogen, Carlsbad, CA, USA) were run under denaturing conditions for 90 min before being transferred to nitrocellulose membranes (Invitrogen) for 2.5 h at 30 V at 4°C in the Blot Module (Invitrogen). Membranes were blocked in blocking buffer [1× Tris-buffered saline (TBS) supplemented with 1% milk] for 1 h. Rabbit anti-PutA antibodies were suspended in blocking buffer at a 1:667 dilution and blotted for 1 h at room temperature. Horseradish peroxidase (HRP)-conjugated antirabbit secondary antibodies (Pierce Biotechnology, Rockford, IL, USA) were suspended in blocking buffer at a 1:5000 dilution and blotted for 30 min at room temperature. The membrane was then incubated with SuperSignal® West Pico chemiluminescent substrate (Pierce Biotechnology) and visualized after exposure to film (Eastman-Kodak, Rochester, NY, USA).
Results and discussion
A strain with the lac operon under transcriptional control of putA was created for use as the host for making mutants. TEV insertions were then constructed in multiple positions in the putA gene through a two-step method using the λ Red system. The initial step required the insertion of CamR, which includes a strong transcriptional terminator to prevent transcription of the downstream lac operon. The second step replaced CamR with the TEVcs, which was selected for by demanding growth on lactose as a sole carbon source (Figure 1). Three classes of mutants resulted from TEVcs insertions: (i) loss of function mutants that did not produce PutA protein; (ii) mutants that produced PutA, but the TEVcs was not accessible to cleavage by the protease; and (iii) mutants that produced PutA and were cleaved by the protease. The first class of mutants that eliminated PutA production all contained TEVcs insertions near residues predicted to be involved with the enzymatic functions of PutA. The phenotype of these mutants suggest that insertion of a TEVcs in close proximity to specific catalytic residues may interfere with protein folding, leading to degradation of PutA protein. The second class of TEVcs insertion mutants that retained wild-type function but were not cleaved by TEV protease are predicted to be located in buried regions of the protein. For example, based on the TEV cleavage patterns, the insertion sites between residues 140–260 are not surface accessible. Although the initial interpretation of the crystal structure suggested that residues 140–260 were involved in dimerization and DNA binding, that interpretation was later revised to suggest that DNA binding and dimerization reside in the first 44 residues of the protein (11).
The third class of mutants produced full-length PutA that was cleaved after induction of the TEV protease. Two of these mutants were particularly interesting. One TEVcs insertion was placed in a region of the protein that had not yet been crystallized, at residue 1224. This mutant was deficient in proline dehydrogenase activity, as well as β-galactosidase activity, suggesting that the insertion enhances the repressor function of PutA. The TEVcs insertion at residue 1224 insertion also cleaved with induction of the TEV protease (Figure 2). Following cleavage in the presence of proline, both proline dehydrogenase activity and β-galactosidase activity decreased 2-fold. Computer analysis of PutA suggests that residues 1204–1220 may participate in membrane binding, hence the TEVcs insertion at 1224 may disrupt the PutA-membrane interaction. The decrease in membrane association is predicted to simultaneously decrease the enzymatic activity of PutA, which requires interaction with the membraneassociated electron transport chain and enhance repressor function of PutA, which requires dissociation from the membrane (9).

Blots were performed after a 4-h induction of the TEV protease. Wildtype (WT) PutA does not cleave with induction of the TEV protease.
A second example of this third class of TEVcs mutants is the insertion at residue 48. The phenotype of the mutant demonstrated an increase in proline dehydrogenase and β-galactosidase activity in the absence of proline, suggesting the TEVcs insertion diminished the ability of PutA to bind DNA. The mutant was cleaved upon induction of the TEV protease (Figure 2). Upon TEV protease cleavage in the absence of proline, β-galactosidase was expressed constitutively, suggesting the DNA binding capability of PutA was further diminished by cleavage of the TEVcs. Also, as expected for a constitutive mutant due to loss of DNA binding, proline dehydrogenase activity in the absence of proline was elevated 5-fold. The phenotype of the mutant post-cleavage indicates that this domain of the protein plays a role in regulation of putA expression. Gu et al. (11) demonstrated the first 47 residues of PutA are capable of binding DNA and dimerization. The phenotype of the TEVcs insertion at residue 48 provides further experimental evidence that the ribbon-helix-ribbon motif identified in the crystal structure is involved in DNA binding function of PutA.
The TEV approach provides a useful assay for studying protein structure and function in vivo. Previous work has used TEV to characterize SecA from E. coli (8), FhaC from Bordetella pertussis (12), and FtsY from E. coli (13). These proteins were quite tolerant of TEVcs insertions. Other studies have found similar results with insertions of different short peptides. For example, Manoil et al. (14) found that 15 out of 23 five-codon insertions caused proteins to retain partial or full function, while the inactive proteins had insertions near important catalytic residues. Overall, PutA was less tolerant of such insertions (Figure 3); however this may be because the multifunctional nature of PutA may demand communication between different domains of the protein (15).

Sites of the 10 insertions are noted by the numbers above the markers. The three classes of mutants are also distinguished as loss of function (LOF), noncleaved mutants (NC), and cleaved mutants (CLV).
Acknowledgments
We thank Michael Ehrmann and Barry Wanner for their generous donation of strains used in this work.
Competing Interests Statement
The authors declare no competing interests.
References
- 1. . 1989. Molecular genetic analysis of a plant virus polyprotein cleavage site: a model. Virology 171:356–364.
- 2. . 1997. TnTIN and TnTAP: Mini-transposons for site specific proteolysis in vivo. Proc. Natl. Acad. Sci. USA 94:13111–13115.
- 3. . 2003. Structure of the proline dehydrogenase domain of the multifunctional PutA flavoprotein. Nat. Struct. Biol. 10:109–114.
- 4. 1990. Experimental Techniques in Bacterial Genetics. Jones and Bartlett, Sudbury, MA.
- 5. . 1988. A novel suicide vector and its use in construction of insertion mutations: osmoregulation of outer membrane proteins and virulence determinants in Vibrio cholerae requires toxR. J. Bacteriol. 170:2575–2583.
- 6. . 2000. Onestep inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. USA 97:6640–6645.
- 7. . 2002. Construction of targeted single copy lac fusions using λ Red and FLP-mediated site-specific recombination in bacteria. Gene 290:153–161.
- 8. . 1996. Site-specific proteolysis of the Escherichia coli SecA protein in vivo. J. Bacteriol. 178:2986–2988.
- 9. . 1997. Regulation of gene expression by repressor localization: biochemical evidence that membrane and DNA binding by the PutA protein are mutually exclusive. J. Bacteriol. 179:2788–2791.
- 10. 1987. Membrane association of proline dehydrogenase in Escherichia coli is redox dependent. Proc. Natl. Acad. Sci. USA 84:373–377.
- 11. . 2004. Identification and characterization of the DNA-binding domain of the multifunctional PutA flavoenzyme. J. Biol. Chem. 279:31171–31176.
- 12. . 2000. Novel topological features of FhaC, the outer membrane transporter involved in the secretion of the Bordetella pertussis filamentous hemagglutinin. J. Biol. Chem. 275:30202–30210.
- 13. . 2001. Evidence for coupling of membrane targeting and function of the signal recognition particle (SRP) receptor FtsY. EMBO Rep. 2:1040–1046.
- 14. . 2000. Insertion of in-frame sequence tags into proteins using transposons. Methods 20:55–61.
- 15. . 1981. Enzymatic properties of the purified putA protein from Salmonella typhimurium. J. Biol. Chem. 256:9762–9766.



