








Western blotting (immunoblotting): history, theory, uses, protocol and problems

Abstract
Western blotting (immunoblotting) is a powerful and commonly used technique that is capable of detecting or semiquantifying an individual protein from complex mixtures of proteins extracted from cells or tissues. The history surrounding the origin of western blotting, the theory behind the western blotting technique, a comprehensive protocol and the uses of western blotting are presented. Lesser known and significant problems in the western blotting field and troubleshooting of common problems are highlighted and discussed. This work is a comprehensive primer and guide for new western blotting researchers and those interested in a better understanding of the technique or getting better results.
Western blotting, also known as immunoblotting, is one of the most commonly used techniques in molecular biology and proteomics in scientific laboratories around the world today [1]. It is an analytical method used to detect and semiquantify target proteins [2,3]. Western blotting also allows the identification of specific amino acids in post-translationally modified proteins that have been modified in the cell due to physiological changes in both healthy and disease states [4]. Such post-translational modification (PTM) of proteins could be in the form of phosphorylation, ubiquitination, biotinylation, glycosylation, methylation, acetylation, sumoylation, nitration, oxidation/reduction, nitrosylation, as well as other types [5].
Although some researchers have criticized western blots and suggested they may not be as reliable as previously assumed, it remains an efficient and powerful technique that can be utilized to identify proteins and accurately quantify relative protein levels [6]. One of the main arguments for western blotting being unreliable is due to poorly characterized antibodies [7]. Researchers have bought commercial protein-detection kits for a specific protein that were later found to be targeting a different protein [8]. However, using validated antibodies, following appropriate experimental procedures and determining the linear and quantitative dynamic range for each target protein under the given experimental conditions make it possible to achieve a successful, reproducible and semiquantitative or quantitative application of western blotting [9]. The theory of western blot, a detailed protocol link, the most common western blotting problems, as well as solutions, troubleshooting tips for common issues and future perspectives are discussed.
History of western blot application in bioscience
The western blot technique was first introduced over 42 years ago (in 1979). Since then, this technique has been mentioned in the abstracts, keywords and titles of more than 400,000 PubMed-listed publications [3]. This highlights the importance of the western blot technique standing the test of time after its first use and its current use in bioscience laboratories today. It is important to understand the history behind the western blot technique to appreciate why it was developed.
In 1807, Ferdinand Frederic Reuss, a physicist at Moscow State University, found that particles moved toward the positive electrode when electricity was passed through a glass tube with clay and water [10]. In 1955, Oliver Smithies separated human tissue extracts using starch gel electrophoresis, which allowed genomic diversity to be estimated at the level of proteins [11]. What is now referred to as western blotting has more than two parents due to multiple research groups working on a similar technique [3]. At least three scientific publications are central to the origin of western blotting. George Stark's laboratory published a method that utilized diffusion for transferring protein to membranes on July 1, 1979 [12]. The Stark's laboratory publication used capillary force for the transfer, a polyacrylamide/agarose gel mix for the separation matrix, diazobenzyloxymethyl paper as the membrane and 125-iodine (I)-labeled protein A to allow detection [12]. The Stark's laboratory was at the forefront of blotting techniques as in 1977 they developed an RNA blotting technique known as “northern blotting” [13]. In 1979, another work by Towbin et al. employed electrophoretic forces to transfer the proteins instead of capillary transfer. Towbin et al. used SDS-PAGE as a separation gel matrix, nitrocellulose membranes as their membrane and primary and secondary antibodies for protein detection [14]. In 1981, 2 years after the publication of the earlier stated publications, W Neal Burnette nicknamed a similar procedure “western blotting” in his publication [15]. He is accredited with being the first to use the name “western blotting” and decided on this name because his laboratory was located on the west coast of the United States. Interestingly, Burnette's article was prepared at the same time the Towbin et al. article was published [12,14]. Burnette's publication was initially rejected; however, Analytical Biochemistry, the journal that initially rejected the manuscript, published the article in 1981 [16]. Burnette's method was comparable to that of Towbin and colleagues in that they both used SDS-PAGE for protein separation and electrophoretic transfer and nitrocellulose as the membrane for immobilization. However, Burnette's method used radiolabeled protein A for detection [3]. It is worth noting that the Towbin et al. publication had over 63,500 citations as of June 2022, substantially more than publications by other inventors of this technique, most likely because their design is the closest to the most commonly used versions of western blotting [3].
Since its development in 1979, some key advances in western blotting include capillary electrophoresis and the development of single-cell western blots [17]. Automation of western blotting was achieved using a capillary electrophoresis system from Protein Simple (CA, USA) [18]. The initial Protein Simple apparatus, Simon, used nanovolume capillaries to separate proteins through a stacking and resolving matrix. Once separated, the proteins are then immobilized to the wall of the capillary tube by photoactivation. Once immobilized, the proteins of interest are identified with primary and horseradish peroxidase (HRP)-conjugated secondary antibodies [18]. An advantage of capillary electrophoresis over the more commonly used (traditional) electrophoresis is that the capillary dimensions (thin and long) result in significantly higher surface area-to-volume ratios, reducing the overheating that occurs at high voltages [18]. Single-cell westerns, which were first described by Amy Herr's laboratory, use a microscope slide with photoactive polyacrylamide gel containing microwells to allow western blotting at the single-cell level [19]. After gel electrophoresis, proteins are immobilized by photoinitiated blotting and subsequent antibody incubations. The advantage of this method is that changes at the level of the single cell can be analyzed, and large amounts of cells could be analyzed relatively quickly (within 4 h).
For such a relatively old technique, one may think western blotting might have been left behind in favor of more modern methods like targeted mass spectrometry, but that is not the case. Publications that mention and apply western blot are significantly greater than publications that apply mass spectrometry for investigating techniques used in protein-related research [3]. Furthermore, compared with other analytical techniques commonly used in protein science, such as ELISA, immunohistochemistry (IHC) and microscopy, western blotting appears to be the most used technique in protein-related publications in the last few years [3]. A search of PubMed on 15 June 2021, showed 66,573 publications using western blotting, 61,845 using IHC and 47,079 using ELISAs between 2019 and 2021 (3 years). These publications included both research and clinical uses of these techniques.
The appeal of western blotting is likely its lower costs and complexity compared with mass spectrometry and other techniques like IHC and ELISA. While the cost of mass spectrometers is prohibitive for most laboratories, the initial costs of equipment for IHC and ELISA techniques are also higher than for western blotting. Another potential factor is that mass spectrometry requires more training and expertise than western blotting to get started. Figures 1 & 2 show the western blotting procedure and possible variations that could occur. The following section will provide in-depth knowledge about the multiple steps employed in western blotting.

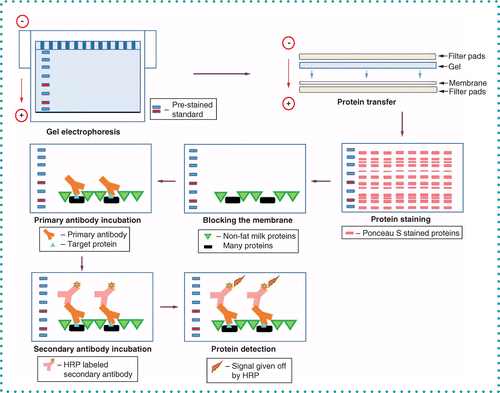
Gel electrophoresis separates proteins based on the size of the protein. Large proteins travel less on gel than smear proteins when an electric charge is conducted across the gel. The protein travels to the positive electrode as the proteins are negatively charged (due to SDS binding). Proteins are then transferred to a membrane that readily binds proteins. Again, an electric charge is used to transfer the proteins from the gel to the membrane. After transfer, the gel is stained to determine the efficiency of the transfer and/or for normalization of semiquantitative results. After destaining, the membrane is blocked and then incubated with the primary antibody. A secondary antibody is used that interacts with the primary antibody. The detection chemistry on the secondary antibody is then used to detect the protein of interest.
BSA: Bovine serum albumin; PBST: Phosphate-buffered saline containing Tween 20; PVDF: Polyvinylidene fluoride; SDS-PAGE: Sodium Dodecyl-sulfate polyacrylamide gel electrophoresis; TBST: Tris-buffered saline containing Tween 20.
HRP: Horseradish peroxidase.
Uses of western blotting
Western blotting is used for a variety of scientific and clinical procedures. The most common uses include detecting a specific protein in a simple or complex mixture of proteins and semiquantification to determine the relative amount of protein in different samples. Western blotting has proven to be invaluable during protein purification or when evaluating the levels of protein expression in cells. The ability of western blotting to provide information about the molecular mass of a protein (i.e., its size) gives it an advantage over antibody-based detection methods such as ELISA and IHC.
Theoretical background of western blotting
Western blotting is a multistep procedure that takes 2–3 days depending on several factors and different protocols used to optimize for the best signal detection. The western blot technique typically involves separating native or denatured proteins based on their molecular weight (size) through gel electrophoresis, transferring these separated proteins to a protein-binding membrane, blocking nonspecific proteins on the membrane with a blocking reagent (typically nonfat milk or bovine serum albumin [BSA]) and subsequent detection of the protein of interest by an antibody specific to the protein of interest [2]. Detecting a signal that reflects antigen/antibody binding can provide semiquantitative or quantitative data about the target protein in simple or complex biological samples such as cell and tissue extracts [20].
Mistakes or significant variations at any process step can influence the result [2]. Several steps, such as the sample preparation method, the amount and source of the primary antibody used and the normalization method used, are essential for reproducible western blot results [2]. Therefore, key steps in this technique must be done using a well-developed procedure to minimize variations and errors that could reduce the reliability and reproducibility of experiments.
Theoretical background of sample preparation
The first important step that should not be overlooked when doing western blotting is sample preparation. To get reproducible results, efficient protein extraction and purification should be carried out using a suitable homogenization method that can efficiently release the intracellular contents of the cell through the rupture of cell membranes [6]. Suitable homogenization methods include ultrasonication, French press, glass-bead milling, mechanical homogenization using Dounce and/or Potter-Elvehjem homogenizers and manual grinding using pestle and mortar, depending on the type of sample that is going to be homogenized [21]. After experiments such as exposure to xenobiotic agents or gene knockdown in mammalian cell culture or animals (e.g., mice, rats, etc.), the cells or tissues from these experiments must be frozen immediately with liquid nitrogen or lysed as quickly as possible to prevent the degradation of proteins by endogenous proteases [6]. Western blotting involves the detection and semiquantification of target protein expression. Therefore, researchers should avoid multiple cycles of repeated freeze/thaw because that could adversely affect some proteins (degradation of some proteins increases with repeated freeze/thaw) [6]. Sample preparation typically starts with lysing the cells or tissues with different lysis buffers such as radioimmunoprecipitation assay buffer (RIPA), Nonidet P-40 (NP-40), Tris-Triton or Tris-HCl buffers with protease/phosphatase inhibitors [22–25]. The choice of lysis buffer should be based on the type of tissue and the location of the target protein inside the cells (i.e., cytosolic, cytoskeletal-bound, membrane-bound, nuclear or mitochondrial) [22,26]. Another important consideration in the choice of lysis buffer depends on whether the primary antibody that will be utilized recognizes native or denatured samples. If the antibody recognizes a nondenatured protein, then buffers with mild nonionic detergents (e.g., NP-40, Triton X-100) or no detergent should be used. The most commonly used components of sample preparation buffers and the function of the components of these buffers are shown in Table 1 (see also [27]). After samples are lysed, they are centrifuged to remove “cellular debris” or fractionated to enrich the sample of interest. However, incorrect results can be obtained if samples are fractionated and a significant amount of the protein of interest is inadvertently discarded as cellular debris during sample preparation [28]. Hence, it is important to determine if the cellular debris contains the target protein when quantifying total homogenates. As such, it is important to determine if the target protein is soluble/cytoplasmic, insoluble or membrane-bound, and which lysis buffer is appropriate for solubilization, fractionation or maintenance of PTM [27].
| Protein location | Buffer | Purpose of components (typical concentrations shown in brackets) |
|---|---|---|
| Whole cell | Nondenaturing, nonionic buffer: NP-40 buffer: 50 mM Tris pH 8.0, 150 mM NaCl, 1% NP-40 or Triton X-100 | Tris-HCl (10–50 mM): buffering of solution (helps maintain stability of proteins) NaCl (50–150 mM): reduces nonspecific binding, maintains ionic strength NP-40/Triton X-100 (0.1–1%): nonionic detergents, solubilizes cytosolic and membrane proteins without denaturing them, permeabilizes membranes and increases solubility of proteins preventing aggregation |
| Denaturing buffer: RIPA buffer: 150 mM NaCl, 1% NP-40 or Triton X-100, 0.5% sodium deoxycholate, 0.1% SDS, 50 mM Tris, pH 8.0 | Sodium deoxycholate (0.1–0.5%): ionic detergent, lyse cells and solubilize cellular and membrane components SDS (0.1–1%): ionic detergent, causes membrane disruption and linearizes proteins by binding to them | |
| Cytoplasmic (soluble) | 20 mM Tris-HCl, pH 7.5 or NP-40 buffer | See above |
| Cytoplasmic (cytoskeletal-bound) | PIPES-Triton buffer: 10 mM PIPES, 50 mM KCl, 1% Triton X-100, 10 mM EGTA, 3 mM MgCl2, 2 M glycerol | PIPES (10 M): buffering of solution KCl (50–150 mM): reduces nonspecific binding, maintains ionic strength EGTA (1–3 mM): inhibition of Ca2+ metalloproteases MgCl2 (1–5 mM): reduces nonspecific binding, together with KCl has a stabilizing effect on some complexes Glycerol (1–10%): stabilizes proteins |
| Membrane-bound | RIPA buffer | See above |
| Mitochondria | RIPA buffer | See above |
| Nuclear | RIPA buffer | See above; note: NP-40 and Triton X-100 cannot lyse nuclear membranes |
| Protein location | Buffer |
It was previously shown by Murphy et al. that the sample enrichment process, which involved centrifugation of muscle tissues, removed an estimated 50% of the myosin present as debris and very likely a similar proportion of the total cellular protein could be lost thereby hindering the ability to quantitatively determine myosin amount [29]. The group also showed that almost two-thirds of another protein of interest, the sarcoplasmic reticulum (SR) Ca2+-binding protein, CSQ2, separated into the debris fraction [29]. Higher CSQ2 in the debris would explain why earlier studies reported a ∼fivefold underestimation of total CSQ2 content in cardiac SR [28]. These examples highlight how fractionation can unintentionally affect protein quantitation and results and emphasize the importance of sample preparation in the western blotting technique. The optimal lysis buffer, centrifugation and/or fractionation should be tested for the protein of interest as some proteins can move between soluble and insoluble fractions due to the treatment used [30].
Theoretical background of gel electrophoresis
Once the protein samples are prepared, the next step is electrophoretic separation. Electrophoresis can be translated to “transport of electricity” from Greek. Gel electrophoresis is one of the major techniques developed to increase selectivity and sensitivity in proteome research [31]. Gel electrophoresis could be carried out using many matrices, including agarose, acrylamide or starch. Acrylamide is the most common matrix for protein gel electrophoresis, partly because the polymerization of acrylamide and bis could be highly regulated to give strong gels with reproducible pore sizes [32]. For protein gels, a ratio of acrylamide to bisacrylamide of 37.5:1 results in a high-resolution protein fractionation method. Different types of polyacrylamide gel electrophoresis exist, with two common types being native PAGE and SDS-PAGE. Native PAGE (often called nondenaturing gels) is used to analyze native proteins. Nondenatured proteins are separated on native PAGE based on net charge, shape and size. Although no heating or addition of SDS occurs to the samples run on native gels, most proteins have a net negative charge in the alkaline buffers used and, as such, these proteins move toward the positive electrode during electrophoresis. Two important features of native PAGE are that enzymatic activities of proteins separated by this method remain intact and most subunit interactions within multimeric proteins are maintained during electrophoresis.
SDS-PAGE has been suggested to be the most widely used technique for analyzing complex mixtures of proteins and separates proteins based on their molecular weight on SDS-PAGE [2,33]. Anionic detergents such as SDS readily react with proteins resulting in negatively charged complexes (the negative charge arising from the SO42- groups of SDS) [34]. The proteins are generally denatured and solubilized by their binding of SDS, and the complex forms a prolate ellipsoid or rod of length roughly proportionate to the protein's molecular weight. SDS strongly binds to proteins at an approximate ratio of one SDS anion for every two amino acids when heated with 0.1% SDS [35]. SDS-bound proteins obtain a net-negative charge proportional to their molecular size and thus travel in the acrylamide gel according to their molecular sizes. The smaller the size of the protein, the faster it travels through the pores of the gel sieve-like matrix of polyacrylamide gel [35]. SDS-PAGE employs the principles of isotachophoresis, which effectively concentrates samples (≥tenfold as measured in the authors' laboratory) from large volumes (within reason) into very small zones, which results in better separation of the different species [36]. The commonly used sample buffer components used to prepare samples for gel electrophoresis and their roles are shown in Table 2, while the common problems associated with gel electrophoresis are shown in Table 3.
| Component | Role |
|---|---|
| Tris-HCl | Used to maintain pH at 8.3 (pH must be adjusted to needed pH) |
| SDS | Aids protein denaturation and provides net-negative charge to protein by uniformly adding SDS to amino acids on the protein |
| 2-mercaptoethanol (β-mercaptoethanol) | Disruption of disulfide bridges aids in protein denaturation |
| Glycerol | Adds density to the buffer, allowing the sample to sink to the bottom of the well |
| Bromophenol blue | Dye to allow user to determine where on the gel the very low-molecular-weight proteins are |
| Gel electrophoresis issue | Possible cause | Possible solution |
|---|---|---|
| Protein bands not properly resolved (too close together) | Inadequate time for sample to run on the gel | Increase gel electrophoresis running time |
| Incorrect pore size used | Use a gradient gel for a broader size range separation of proteins or use a pore size appropriate for the protein being separated | |
| Broad bands | Old sample buffer | Prepare and use a fresh sample buffer |
| Incomplete acrylamide polymerization | Allow longer time for polymerization and use electrophoresis-grade reagents | |
| Band pattern curves upwards (“smiling”) or downwards (“frowning”) in gel lanes | Detergents or solvents in the sample | Remove detergents and solvents by dialysis before sample preparation |
| Proteins overloaded | Decrease amount of protein loaded in each gel | |
| Band pattern curves upward on both sides of gel (“smiling” pattern) | Power used was too high | Lower voltage used to run the gel or use recommended running conditions |
| Insufficient running buffer in the well | Fill inner buffer chamber so that wells are completely covered. Ensure that inner chamber is not leaking into outer chamber | |
| Center of the gel was hotter than sides of the gel. May be due to an incorrect running buffer. | Make a new running buffer and ensure it is mixed well before use. Use chilled buffer or run gel in a refrigerator | |
| Skewed or distorted bands, lateral band spreading | Salt concentration in sample is too high | Remove salt in the sample by dialysis before sample preparation |
| Sample diffusion before starting electrophoresis | Reduce time between loading wells and starting electrophoresis | |
| Less sample buffer than needed used or incorrectly made sample buffer used | Make fresh sample buffer and ensure amount of sample buffer is adequate for volume of sample used | |
| No bands | Power used was too high | Lower voltage used to run the gel or use recommended running conditions |
| Running time was too long | Decrease gel running time | |
| Incorrect % acrylamide gel used | Use a gel with a higher % acrylamide | |
| Too little protein loaded on the gel | Load more protein in each well | |
| Electrophoresis connects were incorrect resulting in sample running upward | Ensure electrophoresis leads are in the correct orientation |
Theoretical background of protein separation
Once protein samples are prepared for gel electrophoresis, the sample is added to the polyacrylamide gel via the wells in the stacking gel. Running buffer is added to the wells before sample addition. The most commonly used running buffers are Tris-glycine running buffers. Other commonly used buffers are acetate, 2-(N-morpholino) ethanesulfonic acid (MES), 3-(N-morpholino) propanesulfonic acid (MOPS), Tris-acetate and Tricine [37,38]. The roles of different components of different running buffers are shown in Table 4.
| Potential component | Role |
|---|---|
| Running buffers | |
| Acetate | Good separation in higher mass range (100–500 kDa) |
| Glycine | Provide glycinate ions to form trailing end during electrophoresis in staking gel |
| MES | Needed to maintain relatively constant pH. Provides MES ions to form trailing end during electrophoresis. MES results in a larger separation at lower mass range (<50 kDa). MES SDS buffer is faster than MOPS SDS buffer due to MES's lower pKa compared with MOPS |
| MOPS | Needed to maintain relatively constant pH. Provides MOPS ions to form trailing end during electrophoresis. MOPS gives better separation at middle to higher mass range. The difference in MES and MOPS ion movement impacts stacking resulting in a difference in protein separation range between MES and MOPS buffers |
| SDS | Helps keeps proteins in a net-negative charge |
| Tricine | Needed to maintain relatively constant pH. Tricine substitutes for glycine in running buffer, providing tricine ions to form trailing end during electrophoresis. Tricine results in more efficient stacking and resolving of low molecular weight proteins. Good for separating low molecular weight proteins (0.5–50 kDa) |
| Tris-HCl | Needed to maintain relatively constant pH. Provides chloride ions that form leading end during electrophoresis in staking gel. Hydrogen ions conduct electricity |
| Transfer buffers | |
| Tris-HCl | Needed to maintain relatively constant pH |
| Glycine | In the absence of methanol, helps prevent gel from swelling |
| Methanol | Prevents gel from swelling during transfer and improves protein binding to nitrocellulose. Removes SDS from proteins allowing better binding to nitrocellulose membranes. Some laboratories no longer include methanol in transfer buffers as results without methanol were not significantly different from those with methanol. Some protocols use 10% ethanol instead of 20% methanol |
| SDS | SDS (up to 0.1%) in transfer buffer increases protein transfer efficiency, especially for large proteins, but may reduce amount of protein binding to membrane. In nitrocellulose membranes with larger pore sizes (0.45 μm), small SDS denatured proteins may go through the membrane |
| CAPS | Needed to maintain relatively constant pH. Recommended for transfer of high molecular weight proteins (>150 kDa) |
For a typical SDS-PAGE system, once electrophoresis starts, both glycinate (from glycine present in the reservoir buffer) and Cl- ions (present throughout the system) begin migrating through the stacking gel [36]. The stacking gel has a lower pH (typically pH 6.8) and Tris-HCl concentration, which enables faster movement of charged particles in the stacking gel. The Cl- ions move at a faster rate than the native or denatured proteins, resulting in what is referred to as the leading ion front (the leading line observed when running the gel). Another front, the trailing front, is formed behind the proteins and is due to the glycinate ions in the buffer. A voltage gradient forms between the glycinate and Cl- ions causing the proteins in the sample loaded to stack (compress) forming a thin layer of proteins. The difference in the pore size between the staking and the resolving gel (due to its smaller pore size) causes increased resistance to the proteins entering the separating gel. The resolving gel also has a higher pH (typically pH 8.8), which is needed to deprotonate the N-terminal amino groups on proteins making them more negatively charged than in the stacking gel [36]. As a result of the higher pH and higher Tris-HCl concentration in the resolving gel, the glycinate and Cl- ions both move faster than the protein ions. The smaller pore size of the resolving gel starts separating proteins based on molecular weight, with smaller proteins moving faster than larger proteins. How fast a charged protein moves (velocity) in a polyacrylamide gel exposed to an electric field can be determined by the equation:
v = E.q/f
v = velocity of molecule
E = electric field (V/cm)
q = net charge on molecule
f = frictional coefficient (depends on the shape and mass of the molecule)
This formula can be used to determine the velocity of proteins during gel electrophoresis but is mostly used to allow a better understanding of the factors that affect the velocity of proteins [39]. The formula shows that proteins will move faster as their net charge increases, as frictional coefficient decreases and as the electric field increases.
Theoretical background of continuous versus discontinuous buffer systems
A continuous buffer system is an electrophoretic system in which the same buffer ions are present throughout the sample, gel and electrode vessel reservoirs at constant pH. The gel is typically made of one continuous acrylamide monomer concentration, and the sample is loaded directly into the gel where separation will occur. The bandwidth is determined in part by the height of the sample load, so samples are usually concentrated and volumes are small for best results [40].
In contrast, discontinuous (isotachophoretic) or multiphasic buffer systems (most used type of gel electrophoresis) comprise two different buffers, such as those with Tris–HCl at the leading electrolyte and Tris-glycine near the terminating electrolyte, in two different gels. The protein sample is added to the stacking gel, which has a lower pH (typically 6.8 in SDS-PAGE gels) and concentration of Tris–HCl (0.125 mol/l). The resolving (separating) gel, which is below the stacking gel, has a higher pH (typically 8.8) and concentration of Tris–HCl (0.375 mol/l). The stacking gel has large pores while the separating gel has smaller pores. The primary ion of the anionic isotachophoretic electrolyte system is the large- and small-pore gel and the anode reservoir is the Cl- ion (from HCl). The Tris-glycine running buffer is added above the stacking gel, which allows the glycinate anion from this buffer to run throughout the gel. Most discontinuous buffer systems have discontinuities of both buffer composition and pH [41].
A discontinuous buffer system is preferable and advantageous over continuous buffer systems because large volumes of dilute protein samples can be applied to the gels, and good resolution of sample components can still be obtained [40]. The better resolution of discontinuous gel systems is due to the two gel layers being made with different buffers (lower concentration and pH in the stacking gel) and larger pore size in the stacking gel [42]. The larger pore size and lower concentration and pH of the gel buffer allow the negatively charged proteins to migrate in between the leading ion (Cl- ions from the stacking gel) and the terminating ion (glycinate from the running buffer). In the resolving gel, due to the smaller pore sizes and the high pH and concentration of the buffer in the gel, the proteins become the terminating ion.
Theoretical background of protein transfer
The next step after electrophoresis is transferring proteins to a membrane. The separated proteins are transferred from the polyacrylamide gel onto microporous surfaces and membranes like cellulose, nitrocellulose, polyvinylidene difluoride (PVDF), cellulose acetate, polyether sulfone and activated nylon [43]. Nitrocellulose is the most commonly used membrane because of its high protein-binding affinity, ability to distinguish between small and large proteins, ability to immobilize proteins and glycoproteins and compatibility with various detection methods (chemiluminescence, chromogenic and fluorescence) [44]. Nitrocellulose is composed of cellulose that is treated with nitric acid, resulting in a brittle cellulose nitrate membrane [45]. Since nitrocellulose is brittle, PVDF is the preferred membrane of choice when the membrane will be reused, such as for reprobing the membrane with a different antibody [43]. PVDF membranes provide better mechanical strength than nitrocellulose membranes, making them appropriate for not just stripping/reprobing but for subsequent sequencing of the amino acids of the proteins on the membrane [46]. Unfortunately, PVDF can sometimes give higher background staining than nitrocellulose, so more optimization to reduce that background is often needed [47].
There are different procedures available to successfully transfer proteins from a gel to a membrane, such as capillary transfer [48], diffusion [49], electroblotting [50] and vacuum blotting [51]. Of these techniques, electroblotting stands out because of its speed and completeness of transfer [50]. The three types of electroblotting are dry electroblotting (dry transfer of proteins to membrane), semidry electroblotting (semidry transfer) and wet electroblotting (wet transfer). Electroblotting is achieved by utilizing an electric field oriented perpendicular to the surface of the gel, causing the charged proteins to move from the gel onto the membrane via the electric current [50]. The membrane is placed facing the positive electrode and the polyacrylamide gel is placed facing the negative electrode. The gel and membrane must be sandwiched to allow efficient transfer. To protect the membrane and gel, filter paper is needed outside the gel and membrane. Fiber pads (sponges) are then added at each end to help create a tight sandwich between the gel and membrane, as any bubbles between the gel and membrane could result in inaccurate results [47]. Immersion of the gel-membrane sandwich (wet transfer) or the addition of transfer buffer-soaked filter paper on either side of the gel-membrane sandwich (semidry transfer) are the most common types of protein transfer for western blotting [50]. For larger proteins, wet transfer is recommended [47].
The most commonly used transfer buffers for western blotting are buffers based on the Towbin system buffer (25 mM Tris, 192 mM glycine, 20% methanol [v/v], pH 8.3 and 0–0.01% SDS) [14]. A variation of the Towbin buffer was developed by Bjerrum Schafer-Nielsen (48 mM Tris, 39 mM glycine, pH 9.2, 20% methanol) and is used for semidry transfer. In some semidry applications, (cyclohexylamino)-L-propanesulfonic acid (CAPS) is substituted for glycine (60 mM Tris and 40 mM CAPS) [52]. The role of the components of the transfer buffer are described in Table 4 (see also [43,53]). It has been found that the lack of methanol in the transfer buffer did not reduce the signal obtained for some proteins [54]. It should also be noted that typical protein transfer is not efficient for proteins with low molecular masses (<10 kDa) and using transfer buffers with a pH that is lower than the isoelectric point of a protein can cause the protein to run backward [50]. Specialized buffers containing CAPS have also been used to transfer proteins with a high isoelectric point to membranes [50].
Theoretical background of membrane blocking
Blocking is the next step after successfully transferring the proteins to the membrane and visualizing them to judge transfer efficiency. This step is important in western blotting because it prevents antibodies from binding to the membrane nonspecifically, reducing background noise in the final result and eradicating false positives [47,50]. To block nonspecific binding, the membrane is incubated in BSA or nonfat dry milk diluted in TBS containing Tween 20 (TBST) or phosphate-buffered saline (PBS) containing Tween 20 (PBST) for 1 h at room temperature [47]. The proteins in milk (mainly casein and whey) or BSA bind to areas of the protein-binding membranes not bound by proteins transferred from the polyacrylamide gel. Hence, milk and BSA prevent nonspecific interactions between the antibodies and the membrane [50]. Nitrocellulose and PVDF are high-affinity protein-binding membranes that bind proteins hydrophobically [55]. BSA and milk proteins may bind weakly to some of the proteins on the membrane that were transferred from gels. Tween 20 reduces interactions between BSA, milk proteins and proteins on the membrane before blocking [50].
Theoretical background of antibody incubation steps
After blocking the membrane to prevent nonspecific binding of proteins and to allow only the detection of the target protein, the next step is to incubate the membrane with primary antibody, wash with TBST or PBST and then incubate with secondary antibody and wash again [50]. Monoclonal and polyclonal antibodies are typically used for western blotting.
The western blotting process relies on two key properties of primary antibodies: specificity, the ability of an antibody to recognize and bind to its target antigen; and selectivity, the preference of an antibody to bind to its target antigen in the presence of a complex mixture of competing proteins [56]. The primary antibody is recognized (interacts with) the secondary antibody. Since the main purpose of the secondary antibody is to enable detection, most secondary antibodies are conjugated to an enzyme, such as alkaline phosphatase (AP) or HRP [57]. Well-characterized antibodies that consistently perform well are therefore essential for reproducible results. Significantly, antibody performance often varies considerably between suppliers and even batches [56]. The antibody concentration that is used is usually based on instructions from the manufacturer, but it is not uncommon to determine a different optimal concentration of antibody. Hence, optimizing the antibody before running all the samples could be a prime determining factor in the assay's sensitivity. Multiple washing with wash buffers such as PBST or TBST is imperative as it minimizes background and removes unbound antibodies. However, the membrane should not be left to wash for a long time (more than 20 min total), as this can also reduce the signal [47].
Theoretical background of antibody signal detection
The final step in the western blot method involves the detection of the probes on labeled antibodies that are bound to the protein of interest. For detection methods, colorimetric, radioactive and fluorescent methods can be used. However, chemiluminescent detection is the most common technique used in western blotting. Enhanced chemiluminescence (ECL) is a sensitive method that can be used for the relative quantitation of the protein of interest [50]. For ECL, the primary antibody binds to the protein of interest and the secondary antibody is tagged with the enzyme HRP, which catalyzes the oxidation of luminol (substrate) in the presence of peroxide to 3-aminophthalate, leading to the emission of light at 428 nm, which is detected by film or by a charged-couple device camera and digital imagers (more sensitive, greater resolution and a larger range of exposures than film) [6,50]. The reaction product produces luminescence, which is relative to the amount of protein detected.
Western blotting using radioactive detection was popular because of its low limit of detection, allowing good signals for weakly expressed proteins. Advancements in both fluorescence and high-sensitivity chemiluminescence now allow safer techniques with low limits of detection [58]. Chromogenic signals can also be used to detect multiplexed signals [59].
Fluorescent dye-conjugated secondary antibodies have been shown to be powerful reagents in the western blotting scheme. There are advantages and disadvantages when using fluorescence instead of chemiluminescence for signal detection. A significant advantage of using fluorescent dye-conjugated secondary antibodies is detecting multiple targets by using fluorophores with nonoverlapping excitation-emission spectra. The somewhat poor performance of fluorophores in the visible range is one possible factor that has limited the use of fluorescence detection for western blotting. However, fluorophores that fluoresce in the infrared spectrum range are as sensitive as chemiluminescence, with the advantage of having a larger linear dynamic range than detection using chemiluminescence [6].
Chromogenic detection works because of chemical reactions triggered by enzymes conjugated with primary or secondary antibodies. The chromogenic substrate is added to a blot previously incubated with an enzyme-conjugated antibody (typically HRP), which converts the substrate to a colored precipitate. Signal development is stopped by simply washing off the substrate. The precipitate on the blot is visible by the eye and can be detected without special equipment as a colored band and does not require excitation by a light source, which is an advantage of chromogenic detection [60,61]. Colorimetric detection is easy to use but may require optimization or additional staining to improve the signal-to-background noise in samples expressing low antigens [27]. 5-Bromo-4-chloro-3-indolyl phosphate (BCIP)/nitro blue tetrazolium (NBT) is a widely used set of chromogenic substrates for the enzyme label AP. Once catalyzed by AP, the product of BCIP further reacts with NBT to produce an insoluble precipitate that is dark blue to purple in color [62].
Radioactive detection requires the use of radioactive probes and the detection involves exposure to x-ray or autoradiography film in the dark [63]. It is relatively rare for this method to be used nowadays, although it was once a popular technique. Some disadvantages of using the radioactive detection method include, but are not limited to, being very expensive, high health and safety risks and the availability of better alternatives.
Practical considerations
Detailed western blotting protocol
A detailed western blotting protocol was created to accompany this publication and can be found at: https://dx.doi.org/10.17504/protocols.io.b5i4q4gw
Transfer conditions
The ability of proteins present in the gel to efficiently transfer to a membrane such as nitrocellulose depends mainly on the molecular mass of the proteins being transferred, the percentage and thickness of the gel used and the type of membrane used [43,64]. In general, protein transfer from the gel to the membrane is more efficient when the gel is thinner and a lower % acrylamide gel is used. If you have trouble transferring your protein from your gel to your membrane, consider using thinner, lower % acrylamide gels [43]. A detailed protocol with formula and instructions for making any % polyacrylamide gels is provided in the Bio-Rad hindcasting polyacrylamide gels bulletin [65].
Nitrocellulose can be prewetted with distilled water or transfer buffer. However, the very high hydrophobic nature of PVDF membranes makes it difficult for proteins to bind to the PVDF membrane during transfer. To help with more efficient transfer, PVDF is prewetted with methanol. Millipore Sigma (MA, USA) introduced a PVDF membrane, Immobilon-E, for western blotting that does not require the prewetting step with methanol. Both nitrocellulose and PVDF membranes are then saturated with transfer buffer and applied to the surface of the gel. An essential aspect of filter paper, gel and membrane physical association is that they should be devoid of air bubbles. Air bubbles prevent the transfer of proteins in the area where the bubble occurs, resulting in artifacts. An effective way to ensure no bubbles are present is to use a pipet to gently roll the surface of the filter paper covering the gel and membrane.
Confirming transfer
Ponceau S, or 3-Hydroxy-4-(2-sulfo-4-[4-sulfophenylazo]phenylazo)-2,7-naphthalenedisulfonic acid sodium salt, can be used to confirm the transfer of the protein on the membranes before blocking and antibody incubation [66]. The first report of using Ponceau S for membrane staining was by Salinovich and Montelaro [67]. After gel electrophoresis and transfer of proteins from the gel to a nitrocellulose or PVDF membrane, Ponceau S staining results in the Ponceau S dye binding to positively charged amino acids and nonpolar regions present on proteins [66]. Ponceau S is a diazo dye that is negatively charged under the conditions (typically 1% acetic acid) used for staining, and when bound to proteins, gives a red/pink stain without significant binding to the membrane itself once quickly washed with water. After imaging the Ponceau S-stained membrane, the stain is easily removed by washing with distilled water or TBS buffer [66]. Ponceau S staining should not be used for nylon membranes, as they are positively charged, as opposed to nitrocellulose or PVDF membranes, which are neutral.
Ponceau S staining is not only used to reveal protein bands but can also be used for western blotting normalization. Normalization of western blotting data is a crucial step that is vital to prevent errors caused by unequal sample loading across different lanes in a gel, inconsistent sample preparation and variations due to other experimental errors [66]. Several papers have shown data that support that total protein normalization is better than housekeeping protein normalization for western blotting normalization [66]. Ponceau S is the most commonly used stain for total protein normalization due to its low cost and fast staining time [66,68]. Based on 2020 prices for Ponceaus S and acetic acid, the authros' group found that the cost to make 100 ml of 0.01% (w/v) Ponceau S in 1% (v/v) acetic acid was less than 20¢ US (not including the cost of water) [66]. Sandler et al. also showed that 1 min of staining time was enough for effective Ponceau S staining [66].
There are many advantages of total protein normalization over housekeeping protein normalization. Most western blotting papers published on PubMed use a housekeeping protein to normalize western blots [66]. The Journal of Biological Chemistry recommended total protein staining as the preferred method for the normalization of western blots seven years ago, while the American Journal of Physiology recommended total protein staining four years ago [69,70]. Therefore, the use of total protein normalization for western blotting is likely to increase substantially over the next few years. While many different recipes for Ponceau S that have been used for membrane staining are available, most recipes use between 0.1% and 2% Ponceau S. The current authors' results showed that a simple Ponceau S stain (0.01% [w/v] Ponceau S in 1% acetic acid [v/v]) gives the same sensitivity of protein detection as all the other Ponceau S formulations containing up to 2% Ponceau S [66].
Another staining method for total protein detection on blots is Stain-Free. When exposed to UV light, tryptophan in proteins are modified with trihalo compounds present in the Stain-Free gel. The modified tryptophan fluoresces strongly under UV light and the intensity of the fluorescence is relative to the amount of protein present. Like Ponceau S staining, Stain-Free has been shown to be a satisfactory loading control. Stain-Free also has the unique advantage of allowing the researcher to view the separation of the proteins on the gel before protein transfer to the membrane. Hence, if the gel ran poorly or had significant artifacts, the process could be stopped at this stage [6]. A comparison of the Stain-Free method and β-actin used as an internal loading control found that Stain-Free total protein normalization was superior to β-actin as a loading control [68].
Blocking conditions
Since milk proteins are not compatible with all detection labels, researchers must choose the appropriate blocking solution. BSA may be preferred over milk as the blocking agent for phosphoprotein antibodies, biotin and AP-labeled antibodies. The major protein component of milk is casein, which is a phosphoprotein. As such when using antiphosphoantibodies, phosphorylated casein could be recognized resulting in extremely high background signals [47]. However, it should be noted that according to Cell Signaling Technology's (MA, USA) in-house tests, nonfat milk worked well for phosphoantibodies [71]. Hence, it may be useful to first determine if the specific phosphoantibody being used works best with milk or BSA (or another blocking buffer). Milk also contains biotin, which inhibits the interaction between avidin (or streptavidin) and biotinylated proteins when using the biotin-avidin detection system [72].
Antibody considerations
It is often overlooked that for all immunodetection steps (such as blocking, antibody incubation, etc.) an adequate volume of liquid and gentle agitation is essential to allow a blot to be evenly exposed to the components without allowing any part of the membrane to dry during the process.
Direct versus indirect antibody detection
While a direct western blot uses one antibody for detection, an indirect western uses two antibodies: the primary antibody that recognizes the target and a labeled secondary antibody (such as HRP-labeled) that interacts with the primary antibody. In the direct detection method of western blotting, a primary antibody is directly conjugated to a reporter enzyme such as AP or fluorescent dye that is used to detect the protein antigen on the membrane after a single incubation step [73]. Hartwell et al. utilized direct immunoblotting for the specific detection of the pathogenic prion protein, PrPSc, a protein that is essential for determining the prion clearance capacity of purification processes for therapeutic proteins [73]. The direct detection method is not as widely used as the indirect detection method despite it being a potentially faster method, due to the advantages of the indirect method, including amplification of signal intensity [27]. Table 5 shows the advantages and disadvantages of direct and indirect detection.
| Direct detection | Indirect detection |
|---|---|
| Advantages | |
| – Typically faster than indirect method as only one antibody is used – Lower chance of a nonspecific signal since a secondary antibody is not used | – Greater signal amplification and stronger signal generation are often possible – Often cheaper to use primary antibody and labeled secondary than to label primary antibody – Allows for changes in detection method after primary antibody is added as secondary antibody could be changed – Allows a wider range of labels |
| Disadvantages | |
| – Labeling primary antibody increases time and cost – Ability of primary antibody to bind its target may be reduced as a result of labeling – Little signal amplification results in weaker signal – Limited flexibility with this detection method | – Increased time due to extra incubation and wash steps required – Increased chance of nonspecific signal from secondary antibody |
Monoclonal versus polyclonal antibodies
Monoclonal antibodies are antibodies produced from a B lymphocyte, namely the fusion with a myeloma cell to yield a hybridoma that secretes the monoclonal antibody, while polyclonal antibodies are a collection of antibodies from multiple different lymphocytes. Since monoclonal antibodies are produced from one B cell, they only react to a single epitope. The main issues regarding monoclonal antibodies revolve around their singular epitope binding. Any changes to the epitope of the target protein may prevent monoclonal antibodies from binding to it. As such, experiments testing for protein presence in specific conditions may affect monoclonal antibody binding if the epitope of the protein is changed due to the condition, whether that be a disease state in the animal or a change to the protein itself. Monoclonal antibodies are also not great for antigen cross-linking experiments and are less sensitive than polyclonal antibodies due to their single-binding epitope [74].
In contrast, polyclonal antibodies recognize multiple epitopes on an antigen, making them great for experiments where there may be some change to the epitope or epitopes of the target protein [74]. These types of antibodies also have a higher specificity than monoclonal antibodies because polyclonal antibodies are composed of many antibodies that target a specific epitope or even multiple epitopes on an antigen [74]. However, polyclonal antibodies are more likely to cause issues with background and cross-reactivity due to the presence of multiple antibodies in the polyclonal antibodies. In general, for best rigor and reproducibility, monoclonal antibodies are preferred over polyclonal antibodies.
Another type of antibody that is becoming more frequently used is genetically engineered antibodies. Recombinant antibodies are a type of genetically engineered antibody. These antibodies are often more reliable than those produced from animals because they are made using the antibodies' specific protein sequence. This allows for less batch variability with these antibodies, allowing for more reproducible experiments [2].
Semiquantification
To help determine how a specific protein or signaling mechanism/pathway responds to a drug treatment or better delineate the mechanism of a disease, western blotting can be employed. Western blots can semiquantify a protein when analyzed with a housekeeping protein or total protein-loaded normalization. The result is semiquantitative because it provides a relative comparison of protein levels, not an absolute measure of quantity [47]. A linear relationship between measurable results and the amount of protein must be established; this correlation is determined by the blotting procedure, primary and secondary antibody binding kinetics, chemiluminescence reaction and image acquisition and processing [75]. When imaging, the determination of the background level is sometimes a problem. Nonspecific background signals are present in all western blots due to several factors including autochemiluminescence of the membranes utilized and the secondary antibodies. For accurate semiquantification of a western blot, it is essential that proper background subtraction is done. The most common background subtraction method seen in the literature entails determining the intensity of an area of the blot without any signal (near the area of interest with a signal) and subtracting this intensity from the target signal intensity [6]. An important aspect of this background subtraction method is that the area that is measured must be the same for the background and the target signal. However, more advanced software allows for more accurate background signal correction and quantifies target bands [76].
Post-translational modifications
Western blotting can be used to detect PTMs, such as the alterations in reversible phosphorylation states of various proteins (phosphoproteins). This is crucial for determining the relative importance of potential signaling mechanisms [27]. The phosphorylation of a protein will alter multiple aspects of its interactions including localization, conformational shape, hydrophobicity and activity [77]. As a result of the development of phosphospecific antibodies, it is now possible to distinguish between different phosphorylated residues (typically Thr, Ser and Tyr) on the same protein of interest [78]. A protein can be phosphorylated at many amino acid residues via different signaling processes. Supplementary Table 1 shows an examination of the importance of semiquantifying the phosphorylated residues that are associated with what is being measured [27,79].
A myriad of other PTMs have important roles in various physiological states, including but not limited to ubiquitination, glycosylation and methylation, and changes may be assessed via western blot. However, other than direct measurements of naturally occurring PTMs, chemically induced PTMs can be incorporated into multiple proteins. For example, using puromycin in cell culture or preclinical models to monitor the translation process. Here, puromycin, a structural analog of aminoacyl tRNAs, is incorporated into newly synthesized proteins that are subsequently probed using an antipuromycin antibody. Measuring the total lane intensity relative to normalization controls provides a semiquantitative assessment of global protein synthesis. Thus, even within the same sample, assessments of signaling cascades (phosphorylation), protein degradation (ubiquitination, using antiubiquitin antibodies) or protein synthesis (puromycin incorporation) may be made, allowing a comprehensive investigation of protein metabolism. Further, PTMs may be assessed by western blotting, such as protein glycosylation. Treating samples with an endoglycosidase (e.g., peptide N-glycosidase F) to remove N-linked glycans results in additional lower molecular weight bands being detected [27]. Hence, western blots can also be used to confirm the presence of some types of protein folding, conformational changes and stability along with solubility.
Another technique that is useful for detecting PTMs (including glycosylation, oxidation, phosphorylation and nitrosylation) on proteins is two-dimensional gel electrophoresis (2DE) [80]. 2DE involves separating proteins according to the net charges on proteins using isoelectric focusing; then, after treatment of the separated samples with SDS, the net charge on the proteins becomes negative and the negatively charged proteins are then separated by SDS-PAGE. An advantage of 2DE is that this technique can simultaneously fractionate, identify and semiquantify proteins when coupled with western blotting or mass spectrometry [80]. 2DE western blots have been used to detect multiple phosphorylated isoforms of an individual protein [81].
Tips & tricks
Common problems with western blots
Sometimes, a western blot will result in visible errors on the membrane. These errors could be weak signal detection, multiple band detection or oversaturation of protein (Table 6).
| Problem | Where problem occurs | Possible cause | Solution |
|---|---|---|---|
| Little or no detection leading to inaccurate results | Sample preparation | Degradation of proteins | Sample was not cold enough during preparation. Keep samples cold during preparation. Avoid multiple freeze/thaw cycles |
| Loss of certain proteins | Use fresh samples instead of frozen samples | ||
| Loss of protein in debris | Optimize sample preparation procedure to ensure that target protein is not in debris that is discarded | ||
| Incorrect or improperly made incubation and washing buffers | Remake all incubation and washing buffers after rechecking concentration of reagents and protocol | ||
| Antibody steps | Unvalidated or inaccurately validated antibody(ies) | Use antibodies that are validated to work under settings being used | |
| Protein loading | Insufficient protein load (from sample protein loss, low expression of protein or poor gel-to-membrane transfer) | Stain membrane after protein transfer to ensure efficient protein transfer. Determine linear range in which signal intensity for target protein is linear | |
| Oversaturated signal leading to inaccurate results | Protein loading | Overloading of sample | Determine linear range in which signal intensity for target protein is linear |
| Antibody steps | Antibody used at too high a concentration | Determine optimal concentration of antibody to use using dot blots or western blotting | |
| Nonspecific band detection | Antibody steps | Unvalidated or inaccurately validated antibody(ies) | Validate antibodies used |
| Blocking step | Insufficient blocking solution or blocking time | Increase blocking time if less than 1 h and increase bovine serum albumin or nonfat milk to 5% | |
| Inaccurate quantification of target protein | Protein loading | Potential overloading or underloading of sample | Determine linear range of signal intensity for target protein. Determine linear range of signal intensity for housekeeping protein |
Problems with preparation
Western blots have many steps where potential errors cause problems with the results, and this begins with the sample preparation. When handling the tissues and cells used for the western blot sample, it is essential to keep them frozen as much as possible. The sample proteins are readily degraded by enzymes present in the tissue or cell, so freezing the samples with liquid nitrogen and keeping them on dry ice is necessary to preserve the sample's proteins. This issue also applies to freezing and thawing the samples. Multiple freeze/thaw cycles interfere with the quality of the protein, so it is best to limit the number of times samples undergo this cycle [2,6]. Using fresh samples usually results in more of certain proteins than after freezing, which is an additional factor to consider when preparing samples. If the target protein is heavily lost or degraded when frozen rather than fresh, it is important to use fresh samples for western blotting [82]. The sample can be treated with protease and phosphatase inhibitors and stored at -80°C to preserve proteins in the sample [82]. In the authors' laboratory, it has been found that treating mouse heart samples with protease inhibitor cocktails and storing them at -80°C prevented the degradation of the proteasome subunits after three years of storage (unpublished results).
Another potential problem encountered in sample preparation occurs when the samples are centrifuged or fractionated. Centrifuging or fractioning samples can help concentrate proteins in the sample but can cause protein loss. Some of the proteins found in high concentrations in cellular debris are myosin or calsequestrin. Removing the cellular debris often results in much lower amounts of these proteins in the sample. Similar issues can result from fractionation, so it is important to verify that centrifugation or fractionation will not cause target protein loss before using these methods [6].
Another potential cause of western blot issues during sample preparation is the buffer used for the sample. The buffer used may not be optimal for the target protein, so it is important to check the type of detergents or reducing reagents used in the buffer to see if they are optimal for the experimental goal (Table 1). However, there are a variety of recipes for this buffer, and many of these recipes do not have well-documented results on their efficacy [2]. The issue could be as simple as the RIPA buffer used followed a recipe from someone who made a mistake documenting the recipe. An easy solution is to remake the buffer based on a RIPA buffer composition that is well documented in the literature.
Some strategies to verify centrifugation and fractionation can be used for isolating organelles and membrane-bound proteins, including using known markers for organelles, or membranes, such as Na/K ATPase for cell membranes [83], or measuring enzyme activities specific to the organelle, such as galactosyl transferase activity in Golgi [84]. To access the homogeneity of the purified organelles, immunofluorescence microscopy or electron microscopy are often carried out [85]. For proteins with unknown or unclear subcellular localization, verification of the protein cellular localization by western blotting and/or immunofluorescence are common techniques [86]. If a protein is present in the cellular debris, then the fractionation method has to be optimized to dissociate the protein [83]. Well-established methods to isolate mitochondria, nucleus and membrane-bound proteins are available [87].
Problems with antibodies
Antibodies are integral in western blots for detecting target proteins in tissues and cells. However, many issues have revolved around the sensitivity, specificity and reliability of commercial antibodies [69]. Antibodies sold by many companies can be of lower quality than advertised and often do not include all information about the antibody, such as information regarding antibody epitopes [2,56,88]. One of the leading causes of these problems is the variation of antibodies due to an animal's immune system [89]. Another reason is that some antibody vendors buy antibodies from other sellers, then relabel and sell those antibodies without providing relevant information about the antibodies. The unreliability of commercial antibodies results in problems such as antibodies nonspecifically binding to nontarget proteins resulting in irreproducible or unusable results [90]. To ensure that the antibody functions as desired, researchers should validate the antibody before experimental use [91]. Problems usually caused by problematic antibodies are weak signal, no signal or nonspecific band detection [47].
There are also online databases that catalog antibodies that researchers have validated. Databases like Antibodypedia (antibodypedia.com), Antibodies-online (antibodies-online.com), the Antibody Registry (antibodyregistry.org), Uniprot (uniport.org) and pAbmAbs (pabmabs.com/wordpress) allow researchers to add or search for antibodies or epitopes that have been validated [7,69,82,88]. If the problem persists after validating the antibody, the protein concentration may be too high. Western blots generally work well with around 10–40 μg of protein per lane loaded into the gel, so lowering the protein concentration can prove beneficial if the sample protein concentration is higher than 40 μg [6].
It is recommended that researchers always test antibodies with positive and negative controls before conducting their experiments. This method ensures that the antibody binds when expected and does not bind to proteins when the target protein is absent [56,69,82,88,90]. For positive controls, use a purified protein sample and/or incubate tissues or cells known to have the target protein with the antibody [69]. For negative controls, a suitable method is to test the antibody using a knockout or knockdown sample, as these are cases where the protein should not be present or present in very low amounts [88]. In addition, incubating membranes with a secondary antibody without incubating the membrane with a primary antibody is a good negative control for any new batch of secondary antibody [69]. If the antibody used in the experiment fails to bind to the positive control, then the antibody is the issue [6,56].
Besides validating antibodies, researchers can also use recombinant antibodies instead of antibodies made in animals for their experiments. As these antibodies are made from their sequence, there is little lot-to-lot variability [56]. Although recombinant antibodies should still be validated to ensure their efficacy for binding as advertised, they allow for more accurate and reproducible results [2].
Concerns with housekeeping proteins
Evidence has shown that housekeeping proteins are not as effective as previously assumed. Housekeeping proteins are used as loading controls for western blots because researchers thought housekeeping protein concentrations were stably expressed throughout various treatments and conditions [92]. However, evidence shows that housekeeping protein concentrations can change under many experimental conditions [69]. This change makes housekeeping proteins more unreliable as a loading control. Researchers will begin relying more on alternative methods like total protein normalization instead. Total protein normalization works by considering the overall intensity of all bands in a lane instead of one housekeeping protein. It accounts for natural variation in the samples, allowing for more accurate protein quantification and analysis [93,94]. Stain-Free western blot gels and Ponceau staining are examples of other ways total protein can be measured as a loading control for western blots [69,95].
Troubleshooting common western blot issues
In weak or no signal cases, the problem is likely due to the antibody (specificity, selectivity or reproducibility), the buffer solution or the antigen [56]. Too little antigen on the membrane can also cause weak or no protein signal. Proteins that are expressed at low levels are more likely to require a longer transfer time or a larger amount of sample [2]. It is also best to use high-quality antibodies for low-expression antigens to increase the likelihood of primary antibody binding. Another potential cause of insufficient antigen on the membrane is that larger proteins can be challenging to properly transfer from a gel to a membrane. Proteins larger than 800 kDa transfer more efficiently using a wet transfer system than other systems, whereas 300 kDa or smaller proteins transfer more efficiently using semidry blotting tools like the Bio-Rad Trans-Blot Turbo machine that can transfer proteins to nitrocellulose membranes in as short a time as 3 min [2].
If a western blot detects nonspecific or multiple bands, it is likely due to the antibody, inefficient blocking or protein concentration. Again, validating the antibody used in the experiment is a valid method of ensuring experimental issues are not caused by problematic antibodies, and protein overloading can also detect multiple bands [96]. Mistakes with blocking the membrane may also cause various bands to appear. Multiple bands could be showing up if the blocking solution is not appropriate for the antibody or if the membrane has not been left in the blocking solution for a long enough time [6].
If the membrane signal is saturated, it is likely due to the overloading of the sample protein. This saturation can generally be solved by either lowering the amount of sample loaded into the gel well or lowering the amount of primary antibody used. Overall, there are many potential causes of problems in western blot experiments. However, understanding common western blot issues can allow researchers to avoid obtaining unusable or irreproducible experimental results.
Conclusion
Western blots will continue to be an essential biochemical method in proteomic research, antibody arrays and the semiquantification of target proteins.
Future perspective
Western blots will continue to be an essential biochemical method in confirming results from proteomic research and antibody arrays and for semiquantification of target proteins. More automation of the western blotting process will occur and improve over the next decade. Research is needed to determine if new automated western blotting machines based on capillary electrophoresis give reproducible results since the normalization process for these methods is not well established. Research is also needed to develop a new group of more easily made, reliable antibodies, such as nanobodies. More robust and cheaper western blots will benefit all scientists.
Western blotting is one of the most powerful and commonly used techniques in scientific laboratories worldwide.
Discussion
Western blotting allows the determination of the relative amounts of a target protein in a complex mixture.
Western blotting allows the identification and quantification of post-translationally modified amino acids in proteins of interest.
Awareness and understanding of common western blot issues allow researchers to avoid irreproducible experimental results.
Supplementary data
To view the supplementary data that accompany this paper please visit the journal website at: www.future-science.com/doi/suppl/10.2144/btn-2022-0034
Author contributions
R Sule: conceptualization and writing: original draft preparation, review and editing; G Rivera: writing: original draft preparation, review and editing; AV Gomes: conceptualization and writing: original draft preparation, review and editing.
Financial & competing interests disclosure
This work was supported by the NIEHS/Superfund Research Program (P42 ES004699). AV Gomes has previously collaborated with Bio-Rad (CA, USA) on western blotting antibodies, reagents and imagers. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
Open access
This work is licensed under the Attribution-NonCommercial-NoDerivatives 4.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
Papers of special note have been highlighted as: • of interest; •• of considerable interest
References
- 1. . Applications of western blot technique: from bench to bedside. Biochem. Mol. Biol. Educ. 49(4), 509–517 (2021).
- 2. . Protein purification and analysis: next generation western blotting techniques. Expert Rev. Proteomics 14(11), 1037–1053 (2017).
- 3. . 40 years western blotting: a scientific birthday toast. J. Proteomics 212, 103575 (2020).
- 4. . The ABCs of PTMs. Nat. Chem. Biol. 14(3), 188–192 (2018).
- 5. . Protein posttranslational modifications: roles in aging and age-related disease. Oxid. Med. Cell Longev. 2017, 5716409 (2017).
- 6. . The necessity of and strategies for improving confidence in the accuracy of western blots. Expert Rev. Proteomics 11(5), 549–560 (2014). • Focuses on best strategies for selecting the best normalization standard, determining the linear range for antibodies and protein stains of the sample of interest and the quality of the primary antibody.
- 7. . Antibody anarchy: a call to order. Nature 527(7579), 545–551 (2015).
- 8. . Translational researchers beware! Unreliable commercial immunoassays (ELISAs) can jeopardize your research. Clin. Chem. Lab. Med. 52(6), 765–766 (2014).
- 9. . A defined methodology for reliable quantification of western blot data. Mol. Biotechnol. 55(3), 217–226 (2013). • Focuses on the importance of data analysis for accurate quantitative western blots.
- 10. . The discovery of electrokinetic phenomena: setting the record straight. Angew. Chemie Int. Ed. 56(29), 8338–8340 (2017).
- 11. . Human genome diversity and its value. In: International Encyclopedia of the Social & Behavioral Sciences. Smelser NJBaltes PB (Eds). Pergamon, Oxford, UK, 6991–6995 (2001).
- 12. . Transfer of proteins from gels to diazobenzyloxymethyl-paper and detection with antisera: a method for studying antibody specificity and antigen structure. Proc. Natl Acad. Sci. USA 76(7), 3116–3120 (1979).
- 13. . Method for detection of specific RNAs in agarose gels by transfer to diazobenzyloxymethyl-paper and hybridization with DNA probes. Proc. Natl Acad. Sci. USA 74(12), 5350–5354 (1977).
- 14. . Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proc. Natl Acad. Sci. USA 76(9), 4350–4354 (1979). •• Introduced the western blotting methodology.
- 15. . “Western blotting”: electrophoretic transfer of proteins from sodium dodecyl sulfate-polyacrylamide gels to unmodified nitrocellulose and radiographic detection with antibody and radioiodinated protein A. Anal. Biochem. 112(2), 195–203 (1981). •• Introduced the name “western blotting.” Paper was initially submitted around the same time as Towbin's 1979 paper.
- 16. . Western blotting: remembrance of things past. In: Western Blotting: Methods and Protocols. Kurien BTScofield RH (Eds). Springer, NY, USA, 9–12 (2015).
- 17. . Single-cell western blotting. Nat. Methods 11(7), 749–755 (2014). •• Introduced single-cell western blotting.
- 18. . The Simple Western™: a gel-free, blot-free, hands-free western blotting reinvention. Nat. Methods 8, v–vi (2011).
- 19. . Single cell-resolution western blotting. Nat. Protoc. 11(8), 1508–1530 (2016).
- 20. . The design of a quantitative western blot experiment. BioMed. Res. Int. 2014, 361590 (2014).
- 21. . A review on macroscale and microscale cell lysis methods. Micromachines (Basel) 8(3), 83 (2017).
- 22. . Methodological considerations for improving western blot analysis. J. Pharm. Toxicol. Methods 61(2), 171–177 (2010).
- 23. . Sample prep for proteomics of breast cancer: proteomics and gene ontology reveal dramatic differences in protein solubilization preferences of radioimmunoprecipitation assay and urea lysis buffers. Proteome Sci. 6, 30 (2008).
- 24. . Advances in preparation of biological extracts for protein purification. Methods Enzymol. 463, 285–303 (2009).
- 25. . Lysis buffer choices are key considerations to ensure effective sample solubilization for protein electrophoresis. Methods Mol. Biol. 1855, 61–72 (2019).
- 26. Characterization of cytoskeleton-enriched protein fraction of the trabecular meshwork and ciliary muscle cells. Invest. Ophthalmol. Vis. Sci. 51(12), 6461–6471 (2010).
- 27. An overview of technical considerations for western blotting applications to physiological research. Scand. J. Med. Sci. Sports 27(1), 4–25 (2017).
- 28. . Important considerations for protein analyses using antibody based techniques: down-sizing western blotting up-sizes outcomes. J. Physiol. 591(23), 5823–5831 (2013).
- 29. . Quantification of calsequestrin 2 (CSQ2) in sheep cardiac muscle and Ca2+-binding protein changes in CSQ2 knockout mice. Am. J. Physiol. Heart Circ. Physiol. 300(2), H595–H604 (2011).
- 30. . An analysis of critical factors for quantitative immunoblotting. Sci. Signal 8(371), rs2 (2015).
- 31. . Improving precision in gel electrophoresis by stepwisely decreasing variance components. J. Pharm. Biomed. Anal. 50(3), 320–327 (2009).
- 32. . Polyacrylamide gel electrophoresis. Science 172(3982), 440–451 (1971).
- 33. . Electrophoresis. In: Encyclopedia of Food Sciences and Nutrition (2nd Edition). Caballero B (Ed.). Academic Press, Oxford, UK, 2055–2062 (2003).
- 34. . The physiological structure and function of proteins. In: Cell Physiology Source Book. Sperelakis N (Ed.). Academic Press, MA, USA, 18–35 (1995).
- 35. . SDS-PAGE and western blotting. In: Diagnostic and Therapeutic Antibodies. George AJTUrch CE (Eds). Humana Press, NJ, USA, 391–405 (2000).
- 36. . SDS polyacrylamide gel electrophoresis of proteins. In: Proteins. Walker JM (Ed.). Humana Press, NJ, USA, 41–55 (1984).
- 37. . Tris-acetate polyacrylamide gradient gels for the simultaneous electrophoretic analysis of proteins of very high and low molecular mass. Methods Mol. Biol. 1855, 269–277 (2019).
- 38. . Tricine-SDS-PAGE. Methods Mol. Biol. 1855, 151–160 (2019).
- 39. . Rules relating electrophoretic mobility, charge and molecular size of peptides and proteins. J. Chromatogr. B. Biomed. Sci. Appl. 699(1–2), 133–147 (1997).
- 40. . Evaluation of commonly used electrophoretic methods for the analysis of proteins and peptides and their application to biotechnology. Analytica Chimica Acta 383(1), 47–60 (1999).
- 41. . Electrophoresis proteins. In: Encyclopedia of Analytical Science (2nd Edition). Worsfold PTownshend APoole C (Eds). Elsevier, Oxford, UK, 460–471 (2005).
- 42. . One-dimensional gel electrophoresis. Methods Enzymol. 463, 497–513 (2009).
- 43. . Western blotting: an introduction. Methods Mol. Biol. 1312, 17–30 (2015).
- 44. . Chemical activation of nitrocellulose membranes for peptide antigen-antibody binding studies: direct substitution of the nitrate group with diaminoalkane. Electrophoresis 14(1), 860–865 (1993).
- 45. . Immobilon-P transfer membrane: applications and utility in protein biochemical analysis. BioTechniques 9(Suppl. 6), 788–805 (1990).
- 46. MStern blotting-high throughput polyvinylidene fluoride (PVDF) membrane-based proteomic sample preparation for 96-well plates. Mol. Cell Proteomics 14(10), 2814–2823 (2015).
- 47. . Western blot: technique, theory, and trouble shooting. N. Am. J. Med. Sci. 4(9), 429–434 (2012).
- 48. . Western blotting using capillary electrophoresis. Anal. Chem. 83(4), 1350–1355 (2011).
- 49. . Diffusion blotting: a rapid and simple method for production of multiple blots from a single gel. Methods Mol. Biol. 1312, 73–76 (2015).
- 50. . The basics of western blotting. Anat. Rec. 295(3), 369–371 (2012).
- 51. . Other notable protein blotting methods: a brief review. Methods Mol. Biol. 1312, 487–503 (2015).
- 52. . Buffer systems and transfer parameters for semidry electroblotting with a horizontal apparatus. In: Electrophoresis' 86. Dunn MJ (Ed.). VCH, Weinheim, Germany, 315–327 (1986).
- 53. . Influence of SDS and methanol on protein electrotransfer to immobilon P membranes in semidry blot systems. BioTechniques 9(4), 397–398; 400–391 (1990).
- 54. . Optimization of western blotting for the detection of proteins of different molecular weight. BioTechniques 68(6), 318–324 (2020).
- 55. . Mechanism of DNA (Southern) and protein (western) blotting on cellulose nitrate and other membranes. J. Chromatogr. 391(1), 53–65 (1987).
- 56. . Antibody validation for western blot: by the user, for the user. J. Biol. Chem. 295(4), 926–939 (2020).
- 57. . Which of the commonly used marker enzymes gives the best results in colorimetric and fluorimetric enzyme immunoassays: horseradish peroxidase, alkaline phosphatase or β-galactosidase? J. Immunol. Methods 79(1), 27–37 (1985).
- 58. . Improving the sensitivity of traditional western blotting via streptavidin containing poly-horseradish peroxidase (PolyHRP). Electrophoresis 40(12–13), 1731–1739 (2019).
- 59. . Detection of multiple antigens on western blots. Anal. Biochem. 236(2), 221–228 (1996).
- 60. . A multiple-staining procedure for the detection of different DNA fragments on a single blot. Anal. Biochem. 190(2), 254–258 (1990).
- 61. . Nonstripping “rainbow” and multiple antigen detection (MAD) western blotting. In: Detection of Blotted Proteins: Methods and Protocols. Kurien BTScofield RH (Eds). Springer New York, NY, USA, 287–301 (2015).
- 62. . Enzyme-linked immunospot assay to monitor antigen-specific cellular immune responses in mouse tumor models. In: Methods in Enzymology. Galluzzi LRudqvist N-P (Eds). Academic Press, 457–477 (2020).
- 63. . Evaluation of biological control with experimental methods. In: Handbook of Biological Control. Bellows TSFisher TW (Eds). Academic Press, CA, USA, 225–242 (1999).
- 64. . Methods Biotechnol. John Wiley & Sons, Hoboken, NJ, USA (2016).
- 65. Bio-Rad Laboratories, Inc.. Handcasting polyacrylamide gels. In: Bulletin 6201. Bio-Rad, CA, USA (2014).
- 66. . Ponceau S waste: ponceau S staining for total protein normalization. Anal. Biochem. 575, 44–53 (2019).
- 67. . Reversible staining and peptide mapping of proteins transferred to nitrocellulose after separation by sodium dodecylsulfate-polyacrylamide gel electrophoresis. Anal. Biochem. 156(2), 341–347 (1986).
- 68. . Stain-Free total protein staining is a superior loading control to β-actin for western blots. Anal. Biochem. 440(2), 186–188 (2013).
- 69. . Guidelines for authors and reviewers on antibody use in physiology studies. Am. J. Physiol. Heart Circ. Physiol. 314(4), H724–H732 (2018).
- 70. . Transparency is the key to quality. J. Biol. Chem. 290(50), 29692–29694 (2015).
- 71. . Tech tips video: milk or BSA? Choosing a blocking protein for western blot (2018). https://blog.cellsignal.com/tech-tips-video-milk-or-bsa-choosing-a-blocking-protein-for-western-blot
- 72. . Inhibition of the streptavidin-biotin interaction by milk. Anal. Biochem. 181(2), 318–320 (1989).
- 73. An improved western blot assay to assess the clearance of prion protein from plasma-derived therapeutic proteins. J. Virol. Methods 125(2), 187–193 (2005).
- 74. . Monoclonal versus polyclonal antibodies: distinguishing characteristics, applications, and information resources. ILAR J. 46(3), 258–268 (2005).
- 75. . Improved semiquantitative western blot technique with increased quantification range. J. Immunol. Methods 345(1), 40–48 (2009).
- 76. . A systematic approach to quantitative western blot analysis. Anal. Biochem. 593, 113608 (2020).
- 77. . Protein electrostatic properties predefining the level of surface hydrophobicity change upon phosphorylation. J. Phys. Chem. Lett. 3(8), 973–976 (2012).
- 78. . Serum antibodies that distinguish between the phospho- and dephospho-forms of a phosphoprotein. Nature 299(5885), 734–736 (1982).
- 79. Regulation of 4E-BP1 phosphorylation: a novel two-step mechanism. Genes Dev. 13(11), 1422–1437 (1999).
- 80. Basics and recent advances of two dimensional- polyacrylamide gel electrophoresis. Clin. Proteomics 11(1), 16 (2014).
- 81. Two-dimensional electrophoresis-based characterization of post-translational modifications of mammalian 20S proteasome complexes. Proteomics 8(23–24), 5025–5037 (2008).
- 82. . Considerations when quantitating protein abundance by immunoblot. Am. J. Physiol. Cell. Physiol. 308(6), C426–C433 (2015).
- 83. . Simple and efficient protocol for subcellular fractionation of normal and apoptotic cells. Cells 10(4), 852 (2021).
- 84. . Identification of new Golgi complex specific proteins by direct organelle proteomic analysis. Proteomics 6(12), 3502–3508 (2006).
- 85. Quantitative proteomics analysis of the secretory pathway. Cell 127(6), 1265–1281 (2006).
- 86. Mapping the murine cardiac 26S proteasome complexes. Circ. Res. 99(4), 362–371 (2006).
- 87. . Isolation of endoplasmic reticulum, mitochondria, and mitochondria-associated membrane and detergent resistant membrane fractions from transfected cells and from human cytomegalovirus-infected primary fibroblasts. Curr. Protoc. Cell. Biol. 68, 32721–232733 (2015).
- 88. . Antibodies: friend or foe? Am. J. Physiol. Gastrointest. Liver Physiol. 309(9), G717–G718 (2015).
- 89. . Reproducibility: standardize antibodies used in research. Nature 518(7537), 27–29 (2015).
- 90. . Research antibodies: do not use them to stain your reputation. Am. J. Physiol. Cell Physiol. 309(11), C707–C708 (2015).
- 91. . Ten basic rules of antibody validation. Anal. Chem. Insights 13, 10.177390118757462 (2018). • Best practices for antibody validation.
- 92. Alterations of housekeeping proteins in human aged and diseased hearts. Pflügers Arch. 473(3), 351–362 (2021).
- 93. Total protein staining is superior to classical or tissue-specific protein staining for standardization of protein biomarkers in heterogeneous tissue samples. Gene Rep. 19, 100641 (2020).
- 94. . Use of the REVERT(®) total protein stain as a loading control demonstrates significant benefits over the use of housekeeping proteins when analyzing brain homogenates by western blot: an analysis of samples representing different gonadal hormone states. Mol. Cell. Endocrinol. 473, 156–165 (2018).
- 95. . Tubulin or not tubulin: heading toward total protein staining as loading control in western blots. Proteomics 17(20), 1600189 (2017). • Critical review comparing housekeeping proteins to total protein staining for normalization of western blots.
- 96. Antibody validation. BioTechniques 48(3), 197–209 (2010).



