







The road to avibactam: the first clinically useful non-β-lactam working somewhat like a β-lactam
Abstract
Avibactam, which is the first non-β-lactam β-lactamase inhibitor to be introduced for clinical use, is a broad-spectrum serine β-lactamase inhibitor with activity against class A, class C, and, some, class D β-lactamases. We provide an overview of efforts, which extend to the period soon after the discovery of the penicillins, to develop clinically useful non-β-lactam compounds as antibacterials, and, subsequently, penicillin-binding protein and β-lactamase inhibitors. Like the β-lactam inhibitors, avibactam works via a mechanism involving covalent modification of a catalytically important nucleophilic serine residue. However, unlike the β-lactam inhibitors, avibactam reacts reversibly with its β-lactamase targets. We discuss chemical factors that may account for the apparently special nature of β-lactams and related compounds as antibacterials and β-lactamase inhibitors, including with respect to resistance. Avenues for future research including non-β-lactam antibacterials acting similarly to β-lactams are discussed.
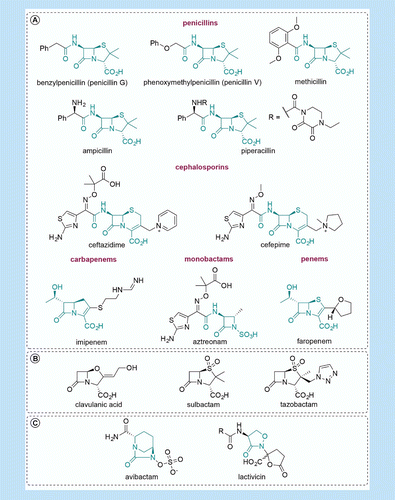
(A) The major classes of β-lactam antibacterials with exemplars from each class with core heterocyclic structures highlighted in green are shown. (B) The clinically used class A β-lactamase inhibitors – clavulanic acid, sulbactam and tazobactam, all contain a β-lactam ring. (C) Avibactam is the first of a new class of β-lactamase, and maybe, penicillin-binding protein inhibitors. Lactivicin is the only naturally occurring clinically relevant penicillin-binding protein inhibitor identified to date which does not contain a β-lactam ring.

(A) Outline mechanism for a bacterical cell wall transpeptidase from a Gram-positive bacterium (PBP: Penicillin-binding protein; GlcNAc: N-acetylglucoseamine; GlcNMur: N-acetylmuramic acid). (B) Comparison of the structure of the D-Ala-D-Ala terminating stem peptide PBP substrate with that of a β-lactam antibacterial. (C) Active site view from a crystal structure of PBP-A (cyan), a PBP from Thermosynechococcus elongatus, the structure of which is related to class A β-lactamases, as acylated by penicillin G (pink) (PDB ID: 2J8Y) [30].

Note in the latter case one or two zinc ions may be present at the active site. Note that in each case the mechanism proceeds via a tetrahedral intermediate. Variations on the mechanisms shown can occur. (C) Active site view from a crystal structure of TEM-1 (cyan), a class A serine-β-lactamase, as acylated by penicillin G (pink) (PDB ID: 1FQG) [63]. (D) Active site view from a crystal structure of NDM-1 (cyan), a B1 metallo-β-lactamase, complexed with hydrolyzed penicillin G (pink) (PDB ID: 4EYF) [64].

(A) Note reactions of the β-lactam inhibitors give multiple acyl–enzyme complexes, some of which are stable to hydrolysis; the precise mechanisms of action may vary from those shown (see main text) [65]. (B) View from a crystal structure of the covalent adduct formed between BlaC (cyan), a serine β-lactamase from Mycobacterium tuberculosis, and clavulanate (pink) (PDB ID: 3CG5) [66].

(A) Compounds with antibacterial activity (5.1 [85], 5.2 [86], 5.3 [87], 5.4 [88], 5.5 [89], 5.6 [90], 5.7 [91] and 5.8 [92]). (B) Examples of inactive compounds (5.9 [93,94], 5.10 [93,94], 5.11 [88], 5.12 [95,96], 5.13 [97], 5.14 [98], 5.15 [99] and 5.16 [100]).

(A) Reversible reaction of a γ-lactam with porcine pancreatic elastase (PPE) [77]. (B) Irreversible reaction of an analogous β-lactam [23]. Note the lactam derived nitrogen in the acyl–enzyme complex of the γ-lactam occupies the space of the ‘hydrolytic water’ as observed in the acyl–enzyme complex derived from a peptide substrate [77]. (C) In (B), the acyl–enzyme complex for the β-lactams has undergone a conformational change; thus, the ester linkage is rotated out of the oxy-anion hole, and the β-lactam carboxylate occupies the space of the hydrolytic water. (D) View from a crystal structure of PPE (cyan) complexed with a monocyclic γ-lactam (pink) (PDB ID: 1QGF) [77]. (E) View from a crystal structure of the acyl–enzyme complex formed between PPE (cyan) and a monocyclic β-lactam (pink) (PDB ID: 1BTU) [23].

(A, B) Note that the bibcyclic lactivicin ring structure is proposed to prevent cycloserine recyclization by opening of the lactone subsequent to that of cycloserine ring [82,83]. (C) View from a crystal structure of the penicillin-binding protein PBP1a (cyan) in complex with the siderophore-derivatised lactivicin shown in (B) (pink) (PDB ID: 4OON) [108].

(A) Outline mode of action of avibactam [143,144]. (B) View from a crystal structure of KPC-2, a serine β-lactamase from Klebsiella pneumoniae, complexed with avibactam (PDB ID: 4ZBE) [143].

Note some avibactam-related compounds are both antibacterials (likely via penicillin-binding protein inhibition) and serine β-lactamase inhibitors [22,139] (9.1 [133], 9.2 [130], 9.3 [129,131–132], 9.4 [145,146], 9.5 [147], 9.6 [147], 9.7 [147], 9.8, 9.9 and 9.10 [148]).

(A) Examples of boronic acids which are proposed as analogues of the tetrahedral intermediates/transition state in β-lactamase/penicillin-binding protein-catalyzed reactions (10.1 [149], 10.2, 10.3 [150], 10.4 [148], 10.5 [151], 10.6 [152], 10.7 [148] and 10.8 [153]). (B) Examples of ‘new’ β-lactam scaffolds also show promise, but they have not been commercially developed (AM-112 (10.9) [154] and LK-157 (10.10) [155]). (C) Examples of natural product derived non-β-lactam type scaffolds which are inhibitors of nucleophilic serine enzymes (trans-lactams (10.11) [101] and a related natural product from Lantana camara (10.11) [156], and the strigolactones (10.13) [157]); derivatives of these could be pursued as penicillin-binding protein/β-lactamase inhibitors.
First draft submitted: 9 April 2016; Accepted for publication: 6 May 2016; Published online: 21 June 2016
Avibactam breaks the β-lactam paradigm
For over 70 years β-lactam compounds have been the most important drugs for treating bacterial infections worldwide [1]. Resistance to the β-lactam antibacterials is a major and growing problem, as is the case for all antibacterials [2,3]. However, the pronounced longevity of the dominance of the β-lactams as antibacterials is remarkable and suggests that there might be underlying molecular reasons for their apparently special nature.
All β-lactam antibacterials target D-ala-D-ala-transpeptidases (penicillin-binding proteins [PBPs]) involved in bacterial cell wall biosynthesis [4–7]. β-Lactams react with a nucleophilic serine in PBPs to form stable acyl–enzyme (ester-linked) complexes which are catalytically inert [5]. Various mechanisms of β-lactam resistance have been identified, including PBP mutations (to give PBPs with low β-lactam affinity) [3,8], modifications to and/or decreased production of outer membrane porin proteins involved in the transport of β-lactams to the periplasm (which lower the effective periplasmic concentration of β-lactams) and increased expression of efflux pumps [9] which efficiently export β-lactams. The most important resistance mechanism to β-lactam antibacterials in Gram-negative bacteria involves β-lactamases, which catalyze the hydrolysis of the β-lactam ring to give inert β-amino acids [3,10–15].
Since the discovery of the penicillins [16], various strategies have been employed to improve the antibacterial properties of the β-lactam antibacterials and to combat resistance mechanisms including via inhibition of β-lactamases [12,17,18]. Until very recently, however, in terms of clinical utility all of these approaches have resulted in β-lactam compounds which are antibacterials, β-lactamase inhibitors or compounds that are both antibacterials and β-lactamase inhibitors [19,20]. Such β-lactam-based compounds include modified penicillins, cephalosporins, monobactams, clavulanic acid and the carbapenems (Figure 1) [5–7,19–21]. Collectively, these β-lactam compounds are a mainstay of modern medicine. The clinical introduction of avibactam (formerly NXL-104, AVE-1330A), a non-β-lactam β-lactamase inhibitor [22], has changed the β-lactam PBP/β-lactamase inhibitor paradigm and, in our view, represents a very major advance in the field. In this overview article, we discuss some of the factors that might contribute to the apparently special nature of β-lactams and outline (bio)chemical work leading to the development of avibactam, and highlight opportunities and challenges which may be stimulated by its clinical introduction.
Although both natural-based and synthetic β-lactams have been shown to have efficacy against other (potential) medicinal chemistry targets (in both micro-organisms and humans) [23,24], all clinically useful β-lactam antibacterials have the same general mode of action, in other words, covalent inhibition of PBPs. PBPs are classified into high- and low- molecular weight proteins [4,7,25–27], the former of which play a crucial role in bacterial cell wall biosynthesis including by catalyzing the cross-linking of peptidoglycan ‘stem peptides’ (Figure 2). The class A, but not class B, high molecular weight PBPs also catalyze the formation of glycan chains in cell wall biosynthesis. The β-lactams inhibit the transpeptidase activity of PBPs by reacting with a catalytically crucial nucleophilic serine residue to form a relatively stable acyl–enzyme complex (Figure 2) [4–7,27–29]. Thus, they target a (near) essential component of enzymes, (some of) which are essential for bacterial survival.
Although nucleophilic serine enzymes are widely distributed in biology, the PBPs (and the mechanistically related serine β-lactamases) are not present in animals, meaning that it may be easier to obtain selective PBP inhibitors than is the case when closely related targets are present in animals and microbes. This task may also be facilitated by the fact that the PBPs are located on the external surface of the cytoplasmic membrane [4]; thus, it is undesirable for β-lactam antibacterials to cross the cell membrane. Bacteria that reside inside human/animals cells, for example, Mycobacterium tuberculosis, are an exception, and β-lactam antibacterials have not been widely used for the treatment of such organisms [31,32]. Although not as widespread as PBPs, there is also evidence that β-lactam biosynthesis is ancient [33–35]. Micro-organisms were likely producing β-lactam antibacterials, long before human/animal use was considered [36]. It would thus seem that the ‘combination’ of β-lactam biosynthesis and PBP inhibition might be one that is particularly ‘useful’ for the β-lactam producers. Given the different types of naturally occurring β-lactams and routes by which they are produced, it is likely that micro-organisms have evolved β-lactam biosynthesis pathways producing PBP inhibitors more than once. The distribution of β-lactam biosynthesis/natural production of PBP inhibitors suggests that β-lactam-mediated PBP inhibition may be one that is not readily prone to the evolution of, at least, facile resistance, consistent with the evidence from the prolonged human use of β-lactam antimicrobials (though it is appreciated that there is now very extensive resistance). As well as interesting medicines/lead compounds, an important aspect of natural product research is identifying useful targets, the β-lactam:PBP ‘combination’ provides an example of how these activities can coincide in a productive manner.
The special nature of β-lactams
The aforementioned considerations raise the question as to whether there is something special about the β-lactam ring itself in terms of PBP and, maybe, β-lactamase inhibition. First, at least the bicyclic β-lactams represent a very densely functionalized low-molecular-weight heterocyclic template which, as revealed by crystallographic analyses, appears highly optimized for PBP inhibition. All the major classes of β-lactam antibacterials (except the clinically used class A serine β-lactamase inhibitors) also contain side chains, which can be, and, have been modified both by nature and medicinal chemists, to improve/modify the properties of the core β-lactam template (Figure 1). Efficient procedures for the exchange of the side chains of the penicillins (at C-6) and cephalosporins (at C-7 and C-3′) have been developed [37–39], and have led to modified penicillins and cephalosporins with improved antibacterial, pharmacokinetics/dynamics and β-lactamase stability properties. The bicyclic cores of all clinically used penicillins and cephalosporins are produced by fermentation, or by modification of fermentation-derived materials; thus, most ‘semi-synthetic’ compounds have relatively low production costs. In contrast, the carbapenems, like the chemically less complex monobactams, are produced by total synthesis (although they were originally generated from natural sources) [40,41]. Although the synthesis of clinically used carbapenems (like meropenem) is presently highly sophisticated, and can be regarded as a triumph of 20th century chemistry, the production cost of (most) carbapenems is considerably higher than most penicillins/cephaloporins; this has likely limited work on the development of new carbapenems (note all clinically used carbapenems have the same [5R,6S]-stereochemistry for their C-6 hydroxyethyl side chain). Overall, however, it is clear that naturally occurring β-lactams have provided a remarkably useful template which can be optimized by side chain (and other) variations.
In order to react with the nucleophilic serine at the PBP active site to form an ester link, β-lactam antibacterials must be sufficiently reactive acylating agents. However, since the natural substrates of PBPs are chemically ‘normal’ peptides/amides, it would seem that the (normally) increased reactivity of β-lactams with respect to acylation/hydrolysis compared with normal amides is not central to their efficacy as inhibitors [42–46]. Increased acylation reactivity to nucleophiles may, however, compensate for a nonoptimized noncovalent binding prior to the acylation step (see, e.g., [23,47,48]). It may also be that the relative liability of β-lactams to hydrolysis, and hence, a limited lifetime in vivo is useful from a clinical perspective because antibacterials are normally only given for a short period.
Although noncovalent binding of the intact β-lactams and product inhibition could play minor roles in inhibition, the available evidence is that all β-lactam antibacterials principally work by forming a stable acyl–enzyme complex with their PBP targets [5–7,27]. This is also the case for clinically useful inhibition of serine β-lactamases [49–51] (Figure 3). Notably, the three clinically used class A (‘penicillinase’) serine β-lactamase inhibitors are themselves all bicyclic β-lactams (clavulanic acid, sulbactam and tazobactam) [49–51], a further example of the ability of β-lactams to inhibit nucleophilic serine enzymes. These compounds are not potent antibacterials, hence are used in combination with penicillin, for example, clavulanic acid and amoxicillin in the Augmentin™ formulation (GlaxoSmithKline, UK). This may at first seem counter-intuitive since β-lactamases bind and efficiently catalyze the hydrolysis of many β-lactam antibacterials. However, β-lactamases are optimized to bind β-lactam antibacterials. Moreover, the acyl–enzyme complexes formed from all three of the clinically useful class A serine β-lactamase inhibitors undergo fragmentation [52–54] to form electrophiles (imines or aldehydes), which can then either react with the water normally involved in acyl–enzyme ester hydrolysis (hydrolytic water) or block access of water to the ester (including via formation of cross-linked species) (Figure 4). Evidence from mass spectrometry [53,54], infrared/Raman spectroscopy [55–57] and crystallographic analyses [58,59] have revealed that in each of these cases multiple inhibitor species are formed following fragmentation of the initially formed acyl–enzyme complex. Evidence, although less definitive, has also been accrued for the formation of more than one acyl–enzyme complex in the reaction of β-lactam antibacterials (e.g., penicillin, ceftaroline) with PBPs [60–62].
The formation of more than one enzyme-inhibitor complex may, in part, rationalize why resistance to β-lactam antibacterials via PBP/β-lactamase mutations has been apparently rather limited. In this regard, the Tipper–Strominger hypothesis [67], in other words, that β-lactam antibacterials mimic the conformation of the D-ala-D-ala component of the PBP bacterial cell wall substrate, may be relevant [68], in that in order to get to the stage where reaction to the acyl–enzyme complex can occur the β-lactam antibacterial must efficiently compete with the natural substrate for binding [69]. Following from biophysical evidence that some nucleophilic serine enzymes may bind their substrates in a high-energy conformation which strains the amide bond [70–72], a refinement of the Tipper–Strominger hypothesis suggests that (some) β-lactam antibacterials may mimic a strained conformation of the D-ala-D-ala entity (Figure 2) [73–75]. Thus, (at least some) β-lactam antibacterials may be highly optimized to bind both noncovalently and covalently; following noncovalent binding, they undergo covalent reaction to form an initial acyl–enzyme complex which at least, in some cases, can undergo either covalent fragmentation [52,53,76] or conformational rearrangement [23,52,55–57,59,62,77] to form (a) more stable complexes. In this scenario, one role of covalent inhibition is to maintain binding of the inhibitor to the enzyme while it is undergoing fragmentation/conformational changes that optimize inhibition/minimize resistance. At present, such ‘mechanism-based’ inhibitors appear to be more common in natural product based medicines (in addition to the β-lactams, a nice example is artemisinin [78,79]) than the outputs of modern medicinal chemistry, which are often optimized for strength of (normally noncovalent) binding, a strategy which if followed without due consideration, might be one which invites rapid development of resistance.
Reversibly binding inhibitors
A further consideration with respect to β-lactam mediated inhibition of nucleophilic serine enzymes, and one relevant to inhibition by avibactam, concerns the reversibility and formation of the acyl–enzyme complex. To date, the available evidence indicates that reactions that form the acyl–enzyme complexes from β-lactams are irreversible. In contrast, work using serine proteases has revealed that appropriately functionalized γ-lactam analogues of β-lactam inhibitors can react reversibly [42,48,77], consistent with knowledge in synthetic chemistry that γ-lactams are (normally) more easily formed than β-lactams in solution. Although considerations other than the reversibility of acyl–enzyme complex formation apply, including the stability of the acyl–enzyme complex with respect to hydrolysis (and as indicated above some β-lactams are optimized in this regard), this work indicates that if the acyl–enzyme can be stabilized compared with the starting γ- (or other non-β-) lactam then, potent non-β-lactam inhibitors of nucleophilic serine enzymes might be possible.
In fact, work on non-β-lactam analogues of the β-lactams had begun in the early days of structure activity studies on the β-lactam antibacterials, with analogues of the penams [80,81]. In the mid-1980s, this work led to the identification of 5,4- and 5,5-lactam ring systems, of which some of the penem analogues displayed antibacterial activity (Figure 5), though the molecular mechanisms underlying the structure activity studies were not apparent at the time. At least in some cases, the antibacterial activity was associated with the increased reactivity of the unsaturated, compared with the saturated, γ-lactams (which as indicated above may be only one factor) [42,77,80–84].
Following the demonstration that γ-, but not β-, lactams can react reversibly with nucleophilic serine enzymes (Figure 6) [42,77], various γ-lactam inhibitors of nucleophilic serine enzymes have been developed, at least some of which derived from the idea of reducing recyclization of the acyl–enzyme complex to the γ-lactam, by reducing the availability of electron density on the nitrogen in the acyl–enzyme complex, for example, by delocalization, with an aromatic ring system [47]. In related work, bicyclic lactam derived ‘trans-lactam’ inhibitors, ultimately derived from more complex natural products, were developed as potent inhibitors of nucleophilic serine proteases [101–104] (though these have not been developed as PBP/serine β-lactamase inhibitors).
Lactivicin
Perhaps, however, the most interesting example of a non-β-lactam acylating inhibitor comes from nature. In 1987, natural product work led to the isolation of lactivicin [105,106], the first and, to date, only naturally occurring non-β-lactam PBP acylating PBP/serine β-lactamase inhibitor. Lactivicin contains a cycloserine ring, in other words, a γ-lactam-related motif, and is fused via its ring nitrogen to a γ-lactone. Both lactivicin and phenoxyacetyl-lactivicin are active against clinically isolated penicillin-resistant Streptococcus pneumoniae strains [107]. Recent efforts to conjugate siderophore mimics to lactivicin have resulted in the broadening of the scope of lactivicin activity to include clinically relevant Gram-negative strains, such as Pseudomonas aeruginosa [108]. Like the β-lactams, lactivicin and its analogues [106,109] irreversibly inhibit PBPs (Figure 7) [82,83,110,111]. Studies, including by crystallography, reveal that the lactivicins react with the nucleophilic serine of PBPs/serine β-lactamases to give an initial acyl–enzyme complex, but that release of the β-lactam derived nitrogen long pair results in opening of the γ-lactone ring to give the crystallographically observed species [82,83]. This secondary fragmentation, which is related to those occurring with some cephalosporins and the class A serine β-lactamase inhibitors (e.g., clavulanic acid), is proposed to stabilize the acyl–enzyme complex [52,76,112–114].
It is also notable that nonacylating inhibitors of PBPs/serine β-lactamases have been developed, including ‘transition state analogue’ based inhibitors, such as boronic acids, some of which show activity in cells [104,115–123] (Figure 10). However, these have not yet reached clinical utility, though some display promise, and are beyond the scope of this article which focuses on mechanistic aspects of acylating inhibitors (see [124–126] for relevant work on boronic acids and related compounds).
Avibactam & DBOs
Avibactam ([2S,5R]-2-carbamoyl-7-oxo-1,6-diazabicyclo[3.2.1.]octan-6-yl hydrogen sulfate) is the prototype of a new type of clinically useful serine β-lactamase and in the future, maybe, PBP/other nucleophilic enzyme inhibitor. Avibactam contains a diazabicyclo[3.2.1]octane (DBO) heterocyclic core structure (Figure 8). The concept of avibactam has its origins in research at Hoechst Marion Roussel, who proposed that DBOs might act to acylate nucleophilic enzymes in a manner analogous to β-lactams [22]. Due, in part, to commercial factors, DBO-based inhibitors were subsequently pursued in Rhône Poulenc and Sanofi Aventis [127], Novexel, and then AstraZeneca [22,128]. These studies ultimately resulted in the clinical development of avibactam, with modified versions of the pioneer DBO compounds being pursued by several companies (e.g., Merck, Novexel) [129–133]. Avibactam has no useful intrinsic antibacterial activity, but is a potent inhibitor of class A, class C and, some, class D serine β-lactamases. It is the only DBO to be approved for clinical use – in combination with a β-lactam antibacterial (Avycaz, ceftazidime-avibactam) [22,134–138]. Interestingly, some DBO derivatives (Novexel) also display antibacterial activity [139,140]. Notably, NXL-105, which belongs to a tricyclic series, exhibits activity against P. aeruginosa, which is likely due to PBP inhibition [133]. Further, some new DBO analogues [22,141,142], such as FPI-1602 (Fedora) (Figure 9), are reported to possess PBP inhibitory activity, in addition to serine β-lactamase inhibitory activity against class A and some class D enzymes [22].
Avibactam mode of action
Detailed kinetic and crystallographic studies have been reported for the interaction of avibactam with class A [158] (TEM-1 [159,160], CTX-M-15 [161], SHV-1 and KPC-2 [143]), class C (P99 [160] and AmpC) and class D (OXA-10, -24 and -48) [162] serine β-lactamases [158]. These studies reveal that, with the isolated enzymes, avibactam has a comparable or better inactivation efficiency for both class A and class C serine β-lactamases compared with the classical β-lactam based class A β-lactamase inhibitor clavulanic acid or tazobactam [160], at least in part, due to the additional stabilization of the carbamoyl–enzyme complex from interactions with polar residues in the active site [160]. Biophysical and kinetic analyses employing mass spectrometry, infrared spectrometry and crystallography reveal that avibactam reacts with the nucleophilic serine to form a relatively stable carbamoyl–enzyme complex [143,161]. In contrast to the fragmentation reactions that β-lactam-based serine β-lactamase inhibitors (e.g., clavulanic acid) can undergo after acyl–enzyme formation [54,112–114] (Figure 4), the available evidence is that, following regioselective ring opening of the 5-membered cyclic urea group of avibactam, a single (major) stable carbamoyl–enzyme complex is formed (Figure 8) [144].
As revealed by a crystal structure of the CTX-M-15-avibactam complex [161], the nitrogen of the N-sulfate group of the ring opened avibactam and a structural water are close to the ester link with the nucleophilic serine (Ser-70); the complex is apparently stabilized by interactions with the Lys-73:Ser-130 and Glu-166:Asn-170 ‘dyads’ which are important in catalysis. The primary amide of avibactam is positioned to interact with the proposed ‘hydrolytic’ water. The avibactam sulfate is positioned to interact with, in a manner similar to binding of the carboxylate of β-lactam substrates (as well as the sulfate of aztreonam), conserved polar residues in the active site [51,161]. These interactions appear to be optimized for binding of the ring opened form of avibactam, and are consistent with the observed long-lived nature of the avibactam complex (t1/2>7 days compared with 5 h for the tazobactam-derived complexes) [141,144,160]. Avibactam is more efficient than the β-lactam based β-lactamase inhibitors in terms of the number of molecules required to inhibit each β-lactamase molecule [158], since it requires only 1–5 avibactam molecules per β-lactamase, compared with the β-lactam inhibitors which require 10s–100s of molecules to completely inhibit a β-lactamase (due to hydrolysis competing with inhibition) [160,163]. Although the number of molecules of DBO required to completely inhibit the β-lactamase may vary with the structure of the DBO and the particular serine β-lactamase, the available evidence is that DBOs appear to be, in general, more efficient than the β-lactam based β-lactamase inhibitors (i.e., clavulanic acid, tazobactam and sulbactam) in this regard [144,163].
Interestingly, and consistent with earlier work on γ-lactam analogues of β-lactams [48], avibactam has been shown to react reversibly with, at least some, serine β-lactamases (e.g., CTX-M-15) [161,164]. Given the DBO core of avibactam, the observation of recyclization is remarkable (though note the nucleophilicity of the nitrogen involved in the reversal reaction is enhanced via the presence of an alpha-effect nucleophile). The reversible reaction of the avibactam carbamoyl-enzyme complex contrasts with the predicted (based on available evidence) irreversible reaction of lactivicin [82], where subsequent ring opening of the lactam linked spirocyclic lactone hinders recyclization of the lactam ring [161]. Hydrolysis of the avibactam derived carbamoyl-enzyme complex can occur, as shown by work on KPC-2 β-lactamase [143] (the avibactam carbamoyl-enzyme complex of which is more prone to hydrolysis than that of CTX-M-15 [161]), through a mechanism which likely includes the initial loss of the N-sulfate via an E1 type elimination to give an imine, which may be hydrolyzed to a ketone, prior to hydrolysis of the carbamoyl-enzyme complex [165]. Time will likely tell, however, whether the formation of a (predominantly) single enzyme–inhibitor complex, rather than multiple complexes, as observed with some β-lactams, will prove an Achilles heel for DBOs with respect to resistance.
Avibactam was approved for clinical use in combination with ceftazidime [138,166] in the USA in 2015 for treatment of complicated urinary tract and intra-abdominal infections for patients with limited or no alternative treatment options. Other combinations of avibactam [167,168], including with ceftaroline [169,170] and aztreonam [171], are in development. The aztreonam–avibactam combination is of interest [137], given that aztreonam is the only β-lactam antibacterial/serine β-lactamase inhibitor nonsusceptible to inactivation by the class B metallo-β-lactamases (MBLs) [14,172] (Figure 3). Derivatization of the primary amide functionality in avibactam has led to the development of new DBO analogues [22], such as 9.1 (NXL-105) [133], 9.2 [130], 9.3 (MK-7655) [129,131–132] and 9.4 (OP0595) [145,146] (Figure 9), some of which have antibacterial activity. In vitro studies have been carried out on these DBO-based compounds, both independently and in combination with various types of β-lactam antibacterials, including penicillins (piperacillin), cephalosporins (cefepime) and carbapenems (imipenem and meropenem) [173–178].
Future perspective
As the first clinically useful non-β-lactam β-lactamase inhibitor, avibactam represents a major breakthrough in the β-lactamase/PBP antibacterial field. The effort required to develop a commercially viable and clinically useful DBO should not be underestimated; the first related patent on DBOs inhibitors was filed by Hoechst Marion Roussel in the mid-1990s [22]. Avibactam, like the carbapenems [40,41], is produced by total synthesis [128] and developing a commercially viable multistep route to prepare avibactam [128] is a considerable achievement. It is to be hoped that the successful synthetic chemistry required to make avibactam viable will inspire future investigations to develop new types of β-lactamase/PBP inhibitors. In this regard, the existing literature suggests that there might be other promising inhibitor templates beyond the classical β-lactams and the DBOs. For example, oxapenems [154,179,180], such as AM-112, are potent β-lactamase inhibitors, though their synthesis is also challenging. Further, there is clearly scope for the development of new types of antibacterial-forming acyl–enzyme type complexes, perhaps based on natural products/inhibitors of nucleophilic serine (or nucleophilic cysteine) enzymes, such as lactivicins [111], trans-lactams [101] or compounds operating via an analogous mechanism of action, but which have been developed in other fields, for example, agrochemistry such as strigolactones [157] (Figure 10). Other types of covalent inhibitors also show promise including transition state analogues such as boronates and phosphonates, which have the potential to target β-lactamases and PBPs [115,118–123]. In this regard, patent applications containing cyclic boronates, which potently inhibits both serine β-lactamases and metallo-β-lactamases, are of particular note [181]. Whether such compounds will be useful as the β-lactams is an open question – the predominance of naturally occurring β-lactam inhibitors of PBPs/β-lactamases suggests not. However, synthetic chemistry can create structures not known to be accessed by nature, as exemplified by the DBO core of avibactam. Although the commercial aspects of the antibacterials are very different from the period when β-lactams were pioneered and developed (from 1940 to early 1990s), it just might be that we are on the course for a period of renewed focus on PBP/β-lactamase inhibitors, not just with β-lactams, but also with β-lactam inspired inhibitors such as avibactam. Just as in the first era of β-lactams, if current efforts are to succeed efficiently, it will require considerable investments from commercial, healthcare and academic sectors, including in fields ranging from basic science (including synthesis) to clinical application.
A subjective factor, not mentioned above in discussing possible factors that may account for the special nature of β-lactams, is that the β-lactams (as many other natural products) are chemically ‘interesting’ and challenging molecules in terms of their structures, mechanisms of action and synthesis. Coupled with their medicinal utility, this ‘interest factor’ may have helped to inspire chemists to work in the β-lactam field. We hope the discovery of the clinical utility of the synthetic DBO ring system, as in avibactam, will inspire further efforts to synthesize chemically interesting ring systems for testing as antibacterials. In this regard, it is notable that there are many reactions available to the modern synthetic chemist, for example, many transition metal catalyzed and asymmetric reactions, which were not available in the first era of β-lactam chemistry.
Executive summary
Bacteria have evolved efficient mechanisms of resistance to β-lactam antibacterials, including by production of β-lactamases which catalyze β-lactam hydrolysis.
Avibactam is a breakthrough because it is a broad-spectrum serine β-lactamase inhibitor, which is not itself a β-lactam; avibactam is approved for clinical use in combination with a cephalosporin (ceftazidime).
Like the β-lactam class A serine β-lactamase and penicillin binding protein inhibitors and the lactivicins (the only naturally occurring small-molecule non-β-lactam penicillin-binding protein/serine β-lactamase inhibitors), avibactam has a core ring structure which enables inhibition via a mechanism involving covalent modification of a catalytically important nucleophilic serine residue.
Unlike β-lactam inhibitors, which react irreversibly, avibactam reacts reversibly with its serine β-lactamase targets.
The advent of avibactam should inspire further efforts to identify non-β-lactam penicillin-binding protein/serine β-lactamase inhibitors.
Financial & competing interests disclosure
The authors would like to thank the following for supporting their work on antibacterials: Medical Research Council (MRC) grant MR/L007665/1, the MRC/Canadian grant G1100135, the MRC SWON alliance, the European Union's Horizon 2020 Research and Innovation Programme under the Marie Sklodowska-Curie grant agreement No 657314, the Biotechnology and Biological Sciences Research Council (BBSRC), AstraZeneca, and the Biochemical Society Krebs Memorial Award. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
Papers of special note have been highlighted as: • of interest; •• of considerable interest
References
- 1 Antibiotic use in eastern Europe: a cross-national database study in coordination with the WHO Regional Office for Europe. Lancet Infect. Dis. 14(5), 381–387 (2014).
- 2 . Antibiotics in the clinical pipeline in 2013. J. Antibiot. 66(10), 571–591 (2013).
- 3 . Diversity of the mechanisms of resistance to β-lactam antibiotics. Res. Microbiol. 142(6), 705–710 (1991).
- 4 . Cutting and stitching: the cross-linking of peptidoglycan in the assembly of the bacterial cell wall. ACS Chem. Biol. 2(9), 602–605 (2007).
- 5 . Penicillin-binding proteins and the mechanism of action of β-lactam antibiotics. Annu. Rev. Biochem. 52, 825–869 (1983).
- 6 . The mechanism of action of penicillin and other β-lactam antibiotics. J. Pharm. Belg. 43(2), 107–115 (1988).
- 7 . The mechanism of the irreversible antimicrobial effects of penicillins: how the β-lactam antibiotics kill and lyse bacteria. Ann. Rev. Microbiol. 33, 113–137 (1979).
- 8 . Penicillin-binding proteins and β-lactamases: their effects on the use of cephalosporins and other new β-lactams. Curr. Clin. Top. Infect. Dis. 8, 37–61 (1987).
- 9 . The importance of efflux pumps in bacterial antibiotic resistance. J. Antimicrob. Chemother. 51(1), 9–11 (2003).
- 10 . Alarming β-lactamase-mediated resistance in multidrug-resistant Enterobacteriaceae. Curr. Opin. Microbiol. 13(5), 558–564 (2010).
- 11 . Metallo-β-lactamases: a last frontier for β-lactams? Lancet Infect. Dis. 11(5), 381–393 (2011).
- 12 . Prolonged infusions of β-lactam antibiotics: implication for antimicrobial stewardship. Pharmacotherapy 32(8), 707–721 (2012).
- 13 . The β-lactamase cycle: a tale of selective pressure and bacterial ingenuity. Nat. Prod. Rep. 16(1), 1–19 (1999).
- 14 . The mechanisms of catalysis by metallo-β-lactamases. Bioinorg. Chem. Appl. 2008, 576297 (2008).
- 15 . A variety of roles for versatile zinc in metallo-β-lactamases. Metallomics 6(7), 1181–1197 (2014).
- 16 . The story of penicillin. Bull. Georgetown. Univ. Med. Cent. 8(4), 128–132 (1955).
- 17 . Targeting antibiotic resistance. Angew. Chem. Int. Ed. Engl.
doi:10.1002/anie.201506818 (2016) (Epub ahead of print). - 18 . Targeting metallo-β-lactamase enzymes in antibiotic resistance. Future Med. Chem. 5(11), 1243–1263 (2013).
- 19 . β-lactamase inhibitor combinations. Med. Clin. North. Am. 79(4), 695–704 (1995).
- 20 . β-lactamase-inhibitor combinations in the 21st century: current agents and new developments. Curr. Opin. Pharmacol. 1(5), 451–458 (2001).
- 21 Current challenges in antimicrobial chemotherapy: focus on β-lactamase inhibition. Drugs 70(6), 651–679 (2010).
- 22 . Diazabicyclooctanes (DBOs): a potent new class of non-β-lactam β-lactamase inhibitors. Curr. Opin. Microbiol. 14(5), 550–555 (2011). •• This article provides an overview of diazabicyclo[3.2.1]octane (DBOs), their devlopment, and highlights their potential as clinical candidates.
- 23 Inhibition of elastase by N-sulfonylaryl β-lactams: anatomy of a stable acyl–enzyme complex. Biochemistry 37(50), 17506–17513 (1998).
- 24 . Monocyclic β-lactam inhibitors of human leukocyte elastase. Tetrahedron 46(7), 2255–2262 (1990).
- 25 . Penicillin binding proteins: key players in bacterial cell cycle and drug resistance processes. FEMS Microbiol. Rev. 30(5), 673–691 (2006).
- 26 . Penicillin-binding proteins and β-lactam resistance. FEMS Microbiol. Rev. 32(2), 361–385 (2008).
- 27 . Mechanism of action of β-lactam antibiotics at the molecular level. Biochem. Pharmacol. 26(23), 2203–2210 (1977).
- 28 . Penicillin-binding protein: the target for β-lactam antibiotics, β-lactamases and their inhibitors. Pharm. Unserer. Zeit. 25(1), 17–27 (1996).
- 29 . β-lactam antibiotic resistance in gram-positive bacterial pathogens of the upper respiratory tract: a brief overview of mechanisms. Microb. Drug. Resist. 1(2), 103–109 (1995).
- 30 . Structure of PBP-A from Thermosynechococcus elongatus, a penicillin-binding protein closely related to class A β-lactamases. J. Mol. Biol. 386(1), 109–120 (2009).
- 31 . Tebipenem, a new carbapenem antibiotic, is a slow substrate that inhibits the β-lactamase from Mycobacterium tuberculosis. Biochemistry 53(22), 3671–3678 (2014).
- 32 . Best drug treatment for multidrug-resistant and extensively drug-resistant tuberculosis. Lancet Infect. Dis. 10(9), 621–629 (2010).
- 33 . Penicillin and cephalosporin biosynthetic genes: structure, organization, regulation, and evolution. Annu. Rev. Microbiol. 46, 461–495 (1992).
- 34 . The enzymology of clavam and carbapenem biosynthesis. Chem. Commun. (34), 4251–4263 (2005).
- 35 . The biosynthesis of β-lactams. In: The Chemistry of β-Lactams. Page MI (Ed.). Springer, The Netherlands, 1–78 (1992).
- 36 . Evolution of isopenicillin N synthase genes may have involved horizontal gene transfer. Mol. Biol. Evol. 7(5), 399–406 (1990).
- 37 . The synthesis of natural β-lactam antibiotics. Contemp. Org. Synth. 1, 417–431 (1994).
- 38 The Chemistry of β-Lactams. Page MI (Ed.). Springer, The Netherlands (1992).
- 39 . β-lactams in the new millennium. Part-I: monobactams and carbapenems. Mini. Rev. Med. Chem. 4(1), 69–92 (2004).
- 40 . Enantioselective syntheses of carbapenem antibiotics. Heterocycles 25(1), 729–806 (1987).
- 41 . The structural aspects of carbapenem antibiotics. Heterocycles 54(1), 497–528 (2001).
- 42 . Reversible acylation of elastase by γ-lactam analogues of β-lactam inhibitors. Bioorg. Med. Chem. Lett. 7(23), 2973–2978 (1997).
- 43 . Hydrolytic stability versus ring size in lactams: implications for the development of lactam antibiotics and other serine protease inhibitors. J. Med. Chem. 43(22), 4328–4331 (2000).
- 44 . The mechanisms of reactions of β-lactam antibiotics. Adv. Phys. Org. Chem. 23, 165–270 (1987).
- 45 . Intramolecular general acid catalysis in the aminolysis of benzylpenicillin. A preferred direction of nucleophilic attack. J. Chem. Soc. Chem. Commun. 6, 298–299 (1979).
- 46 . Alcohol-catalysed hydrolysis of benzylpenicillin. J. Chem. Soc. Perkin Trans. 2(8), 1213–1217 (1991).
- 47 5,5-fused thiophene γ-lactams as templates for serine protease inhibition. Chem. Commun. (12), 1274–1275 (2002).
- 48 . ’pH-jump’ crystallographic analyses of γ-lactam-porcine pancreatic elastase complexes. Biochem. J. 351(Pt 2), 335–340 (2000).
- 49 . Updated functional classification of β-lactamases. Antimicrob. Agents Chemother. 54(3), 969–976 (2010).
- 50 . Three decades of β-lactamase inhibitors. Clin. Microbiol. Rev. 23(1), 160–201 (2010).
- 51 . β-lactamase inhibitors. Drug. Resist. Update 3(2), 109–125 (2000).
- 52 High resolution crystal structures of the trans-enamine intermediates formed by sulbactam and clavulanic acid and E166A SHV-1 β-lactamase. J. Biol. Chem. 280(41), 34900–34907 (2005).
- 53 Mechanism of inhibition of the class A β-lactamases PC1 and TEM-1 by tazobactam. Observation of reaction products by electrospray ionization mass spectrometry. Biol. Chem. 275(35), 26674–26682 (2000).
- 54 . Inhibition of TEM-2 β-lactamase from Escherichia coli by clavulanic acid: observation of intermediates by electrospray ionization mass spectrometry. Biochemistry 35, 12421–12432 (1996).
- 55 “Mind the gap”: Raman evidence for rapid inactivation of CTX-M-9 β-lactamase using mechanism-based inhibitors that bridge the active site. J. Am. Chem. Soc. 137(40), 12760–12763 (2015).
- 56 . Following the reactions of mechanism-based inhibitors with β-lactamase by Raman crystallography. Biochemistry 42(46), 13386–13392 (2003).
- 57 Why clinically used tazobactam and sulbactam are poor inhibitors of OXA-10 β-lactamase: Raman crystallographic evidence. Biochemistry 47(13), 4094–4101 (2008).
- 58 The importance of the trans-enamine intermediate as a β-lactamase inhibition strategy probed in inhibitor-resistant SHV β-lactamase variants. ChemMedChem 7(6), 1002–1008 (2012).
- 59 Tazobactam forms a stoichiometric trans-enamine intermediate in the E166A variant of SHV-1 β-lactamase: 1.63 a crystal structure. Biochemistry 43(4), 843–848 (2004).
- 60 . The allosteric site for the nascent cell wall in penicillin-binding protein 2a: an Achilles’ heel of methicillin-resistant Staphylococcus aureus. Curr. Med. Chem. 22(14), 1678–1686 (2015).
- 61 . Penicillin-binding protein 2a of methicillin-resistant Staphylococcus aureus. IUBMB Life 66(8), 572–577 (2014).
- 62 Structural basis for clinical longevity of carbapenem antibiotics in the face of challenge by the common class A β-lactamases from the antibiotic-resistant bacteria. J. Am. Chem. Soc. 120(38), 9748–9752 (1998).
- 63 Molecular structure of the acyl–enzyme intermediate in β-lactam hydrolysis at 1.7 A resolution. Nature 359(6397), 700–705 (1992).
- 64 . New Delhi metallo-β-lactamase: structural insights into β-lactam recognition and inhibition. J. Am. Chem. Soc. 134(28), 11362–11365 (2012).
- 65 . Resistance to β-lactam antibiotics: structure and mechanism based design of β-lactamase inhibitors. Curr. Med. Chem. 9(12), 1145–1165 (2002).
- 66 . Structure of the covalent adduct formed between Mycobacterium tuberculosis β-lactamase and clavulanate. Biochemistry 47(19), 5312–5316 (2008).
- 67 . Mechanism of action of penicillins: a proposal based on their structural similarity to acyl-D-alanyl-D-alanine. Pro. Natl Acad. Sci. USA 54(4), 1133–1141 (1965).
- 68 . The Tipper-Strominger hypothesis and triggering of allostery in penicillin-binding protein 2a of methicillin-resistant Staphylococcus aureus (MRSA). J. Am. Chem. Soc. 137(20), 6500–6505 (2015).
- 69 . Bacterial cell wall synthesis and structure in relation to the mechanism of action of penicillins and other antibacterial agents. Am. J. Med. 39(5), 708–721 (1965).
- 70 . Structural analyses on intermediates in serine protease catalysis. J. Biol. Chem. 281(33), 24024–24035 (2006).
- 71 . Restricting the psi torsion angle has stereoelectronic consequences on a scissile bond: an electronic structure analysis. Biochemistry 54(37), 5748–5756 (2015).
- 72 . A computational evaluation of the mechanism of penicillin-binding protein-catalyzed cross-linking of the bacterial cell wall. J. Am. Chem. Soc. 133(14), 5274–5283 (2011).
- 73 . Conformation of penicillin as a transition-state analog of the substrate of peptidoglycan transpeptidase. J. Mol. Biol. 61(2), 463–469 (1971).
- 74 . Transition state structures of a dipeptide related to the mode of action of β-lactam antibiotics. Pro. Natl Acad. Sci. USA 74(12), 5239–5243 (1977).
- 75 . Crystal structures of complexes between the R61 DD-peptidase and peptidoglycan-mimetic β-lactams: a non-covalent complex with a “perfect penicillin”. J. Mol. Biol. 345(3), 521–533 (2005).
- 76 . Inhibition of TEM-2 β-lactamase from Escherichia coli by clavulanic acid: observation of intermediates by electrospray ionization mass spectrometry. Biochemistry 35(38), 12421–12432 (1996).
- 77 Mechanistic insights into the inhibition of serine proteases by monocyclic lactams. Biochemistry 38(25), 7989–7998 (1999).
- 78 . A new look at an old drug: artemisinin and qinghaosu. Papua New Guinea Med. J. 35(4), 264–269 (1992).
- 79 . Artemisinin and its derivatives: a novel class of anti-malarial and anti-cancer agents. Chem. Soc. Rev. 39(2), 435–454 (2010).
- 80 . γ-lactam analogues of β-lactam antibiotics. J. Antibiot. 44(1), 1–24 (1991).
- 81 The Chemistry Of Penicillin: Report On A Collaborative Investigation By American And British Chemists Under The Joint Sponsorship Of The Office Of Scientific Research And Development, Washington, DC., And The Medical Research Council, London. Princeton University Press, London (1949).
- 82 . Structural basis for the interaction of lactivicins with serine β-lactamases. J. Med. Chem. 53(15), 5890–5894 (2010).
- 83 Structural and mechanistic basis of penicillin-binding protein inhibition by lactivicins. Nat. Chem. Biol. 3(9), 565–569 (2007).
- 84 . Antibacterial properties of the bicyclic pyrazolidinones. J. Antibiot. 43(1), 92–99 (1990).
- 85 Merck & Co: US4428960 (1984).
- 86 . A γ-lactam analogue of penems possessing antibacterial activity. Tetrahedron Lett. 27(30), 3461–3464 (1986).
- 87 . γ-lactam analogues of the penems. Tetrahedron Lett. 27(30), 3453–3456 (1986).
- 88 Molecular modelling of γ-lactam analogues of β-lactam antibacterial agents: synthesis and biological evaluation of selected penes and carbapenem analogues. Tetrahedron 45(7), 1905–1928 (1989).
- 89 γ-lactam analogues of carbapenems. Tetrahedron Lett. 27(30), 3457–3460 (1986).
- 90 . Synthesis of γ-lactam analogues of carbapenems with substituted thio groups at the C-3 position. J. Chem. Soc. Parkin Trans. 1, 2345–2352 (1988).
- 91 . [3.3.0]Pyrazolidinones: an efficient synthesis of a new class of synthetic antibacterial agents. Tetrahedron Lett. 31(20), 2805–2808 (1990).
- 92 . Bicyclic pyrazolidinone antibacterial agents: synthesis of side chain analogues of carbapenems PS-5 and thienamycin. Tetrahedron Lett. 30(1), 1889–1892 (1989).
- 93 . γ-lactam analogues of carbapenicillanic acids. J. Chem. Soc. Chem. Commun. (4), 194–196 (1985).
- 94 . γ-lactam analogues of carbapenicillanic acids. Tetrahedron 42(17), 4879–4888 (1986).
- 95 . γ-lactam analogues of penicillanic and carbapenicillanic acids. Tetrahedron 40(21), 4513–4525 (1984).
- 96 . γ-lactam analogues of penicillanic and carbapenicillanic acids. J. Chem. Soc. Chem. Commun. (5), 250–252 (1983).
- 97 . Access to a homocephalosporin via ring expansion of a homopenicillin sulfoxide. ChemInform 23(3), (1992).
- 98 . Synthesis of novel bicyclic γ-lactam analogue of the 1-oxapenams. Tetrahedron Lett. 30(30), 4019–4020 (1989).
- 99 . Thioaldehydes in cycloaddition reactions. Synthesis of nuclear analogues of pyrazolidnone antibacterial agents. Tetrahedron Lett. 29(40), 5061–5064 (1988).
- 100 . [4.3.0]Pyrazolidinones as potential antibacterial agents. Tetrahedron Lett. 29(50), 6569–6572 (1988).
- 101 Design and synthesis of pyrrolidine-5,5-trans-lactams (5-oxo-hexahydro-pyrrolo[3,2-b]pyrroles) as novel mechanism-based inhibitors of human cytomegalovirus protease. 1. The α-methyl-trans-lactam template. J. Med. Chem. 43(23), 4452–4464 (2000).
- 102 Pyrrolidine-5,5-trans-lactams. 1. Synthesis and incorporation into inhibitors of hepatitis C virus NS3/4A protease. Org. Lett. 4(25), 4475–4478 (2002).
- 103 Design and synthesis of pyrrolidine-5,5′-trans-lactams (5-oxo-hexahydropyrrolo[3,2-b]pyrroles) as novel mechanism-based inhibitors of human cytomegalovirus protease. 4. Antiviral activity and plasma stability. J. Med. Chem. 46(21), 4428–4449 (2003).
- 104 . Reactivity and selectivity in the inhibition of elastase by 3-oxo-β-sultams and in their hydrolysis. Org. Biomol. Chem. 5(24), 3993–4000 (2007).
- 105 . Structure of lactivicin, an antibiotic having a new nucleus and similar biological activities to β-lactam antibiotics. Tetrahedron Lett. 27(51), 6229–6232 (1986).
- 106 . γ-lactam analogues of monocyclic β-lactam antibiotics. Tetrahedron Lett. 28(25), 2883–2886 (1987).
- 107 . Lactivicin – an antibiotic against penicillin-resistant pneumococci. Pharm. Unserer. Zeit. 36(6), 421–422 (2007).
- 108 Siderophore receptor-mediated uptake of lactivicin analogues in Gram-negative bacteria. J. Med. Chem. 57(9), 3845–3855 (2014). • This article highlights new derivatives of lactivicin and its action against Gram-negative bacteria.
- 109 . Synthesis and antibacterial activity of lactivicin derivatives. Chem. Pharm. Bull. 38(1), 116–122 (1990).
- 110 Binding of a non-β-lactam antibiotic to penicillin-binding proteins. Nature 325(7000), 179–180 (1987).
- 111 . Lactivicin, a naturally occurring non-β-lactam antibiotic having β-lactam-like action: biological activities and mode of action. J. Antibiot. 42(1), 84–93 (1989).
- 112 . Mass spectrometric studies on the inhibition of TEM-2 β-lactamase by clavulanic acid derivatives. J. Antibiot. 50(2), 184–185 (1997).
- 113 . Interaction of clavulanate with the β-lactamases of streptomyces albus G and actinomadura R39. Biochem. J. 207(3), 429–436 (1982).
- 114 . Chemical studies on the inactivation of Escherichia coli RTEM β-lactamase by clavulanic acid. Biochemistry 17(11), 2185–2189 (1978).
- 115 . Development of new drugs for an old target: the penicillin binding proteins. Molecules 17(11), 12478–12505 (2012).
- 116 . β-sultams-mechanism of reactions and use as inhibitors of serine proteases. Acc. Chem. Res. 37(5), 297–303 (2004).
- 117 Different transition-state structures for the reactions of β-lactams and analogous β-sultams with serine β-lactamases. J. Am. Chem. Soc. 127(49), 17556–17564 (2005).
- 118 . Use of novel boronic acid transition state inhibitors to probe substrate affinity in SHV-type extended-spectrum β-lactamases. Antimicrob. Agents Chemother. 51(4), 1577–1579 (2007).
- 119 Boronic acid transition state inhibitors active against KPC and other class A β-lactamases: structure–activity relationships as a guide to inhibitor design. Antimicrob. Agents Chemother. 60(3), 1751–1759 (2016).
- 120 Novel insights into the mode of inhibition of class A SHV-1 β-lactamases revealed by boronic acid transition state inhibitors. Antimicrob. Agents Chemother. 55(1), 174–183 (2011).
- 121 Unexpected tricovalent binding mode of boronic acids within the active site of a penicillin-binding protein. J. Am. Chem. Soc. 133(28), 10839–10848 (2011).
- 122 . Crystal structure of Enterobacter cloacae 908R class C β-lactamase bound to iodo-acetamido-phenyl boronic acid, a transition-state analogue. Cell. Mol. Life Sci. 60(8), 1764–1773 (2003).
- 123 . The deacylation mechanism of AmpC β-lactamase at ultrahigh resolution. J. Am. Chem. Soc. 128(9), 2970–2976 (2006).
- 124 Design and exploration of novel boronic acid inhibitors reveals important interactions with a clavulanic acid-resistant sulfhydryl-variable (SHV) β-lactamase. J. Med. Chem. 56(3), 1084–1097 (2013).
- 125 Crystal structures of KPC-2 β-lactamase in complex with 3-nitrophenyl boronic acid and the penam sulfone PSR-3-226. Antimicrob. Agents Chemother. 56(5), 2713–2718 (2012).
- 126 . Structural study of phenyl boronic acid derivatives as AmpC β-lactamase inhibitors. Bioorg. Med. Chem. Lett. 20(11), 3416–3419 (2010).
- 127 Aventis Pharma SA: WO2002010172 (2002).
- 128 AstraZeneca: WO2012172368 (2012).
- 129 Merck: WO2009091856 (2009).
- 130 Merck: WO2012086241 (2012).
- 131 . A concise synthesis of a β-lactamase inhibitor. Org. Lett. 13(20), 5480–5483 (2011).
- 132 Practical and cost-effective manufacturing route for the synthesis of a β-lactamase inhibitor. Org. Lett. 16(1), 174–177 (2014).
- 133 Novexel: WO2008142285 (2008).
- 134 Efficacy and safety of ceftazidime-avibactam versus imipenem-cilastatin in the treatment of complicated urinary tract infections, including acute pyelonephritis, in hospitalized adults: results of a prospective, investigator-blinded, randomized study. Curr. Med. Res. Opin. 28(12), 1921–1931 (2012).
- 135 . Comparative study of the efficacy and safety of ceftazidime/avibactam plus metronidazole versus meropenem in the treatment of complicated intra-abdominal infections in hospitalized adults: results of a randomized, double-blind, Phase II trial. J. Antimicrob. Chemother. 68(5), 1183–1192 (2013).
- 136 . Improving known classes of antibiotics: an optimistic approach for the future. Curr. Opin. Pharmacol. 12(5), 527–534 (2012).
- 137 Activities of NXL104 combinations with ceftazidime and aztreonam against carbapenemase-producing Enterobacteriaceae. Antimicrob. Agents Chemother. 55(1), 390–394 (2011).
- 138 Actavis Inc. Prescribing Information: AVYCAZ (ceftazidime-avibactam) for injection, for intravenous use. www.accessdata.fda.gov/drugsatfda_docs/label/2015/206494.
- 139 . New β-lactam antibiotics and β-lactamase inhibitors. Expert Op. Ther. Pat. 20(10), 1277–1293 (2010).
- 140 . NXL-103, a combination of flopristin and linopristin, for the potential treatment of bacterial infections including community-acquired pneumonia and MRSA. Curr. Opin. Investig. Drugs 11(2), 225–236 (2010).
- 141 Structural and kinetic characterization of diazabicyclooctanes as dual inhibitors of both serine-β-lactamases and penicillin-binding proteins. ACS Chem. Biol. 11(4), 864–868 (2016).
- 142 Enantioselective synthesis and profiling of two novel diazabicyclooctanone β-lactamase inhibitors. ACS Med. Chem. Lett. 5(10), 1143–1147 (2014).
- 143 . Inhibition of Klebsiella β-lactamases (SHV-1 and KPC-2) by avibactam: a structural study. PLoS ONE 10(9), e0136813 (2015).
- 144 Avibactam is a covalent, reversible, non-β-lactam β-lactamase inhibitor. Proc. Natl Acad. Sc. USA 109(29), 11663–11668 (2012). Provides a description of the detailed mechanism of action of avibactam with TEM-1.
- 145 Meiji Seika Pharma Co. Ltd: WO2013180197 (2012).
- 146 OP0595, a new diazabicyclooctane: mode of action as a serine β-lactamase inhibitor, antibiotic and β-lactam ‘enhancer’. J. Antimicrob. Chemother. 70(10), 2779–2786 (2015).
- 147 Structural and kinetic characterization of diazabicyclooctanes as dual inhibitors of both serine-β-lactamases and penicillin-binding proteins. ACS Chem. Biol. 11(4), 864–868 (2016).
- 148 . β-lactamase inhibitors: a review of the patent literature (2010 – 2013). Expert Opin. Ther. Pat. 23(11), 1469–1481 (2013). •• This article reviews the recent patent literature and highlights new β-lactamase inhibitors.
- 149 Fragment-guided design of subnanomolar β-lactamase inhibitors active in vivo. Proc. Natl Acad. Sci. USA 109(43), 17448–17453 (2012).
- 150 . 4,7-Dichloro benzothien-2-yl sulfonylaminomethyl boronic acid: first boronic acid-derived β-lactamase inhibitor with class A, C, and D activity. Bioorg. Med. Chem. Lett. 20(8), 2622–2624 (2010).
- 151 . Structure-based design of a potent transition state analogue for TEM-1 β-lactamase. Nat. Struct. Biol. 3(8), 688–695 (1996).
- 152 Optimizing cell permeation of an antibiotic resistance inhibitor for improved efficacy. J. Med. Chem. 50(23), 5644–5654 (2007).
- 153 Synthesis and SAR of novel benzoxaboroles as a new class of β-lactamase inhibitors. Bioorg. Med. Chem. Lett. 21(8), 2533–2536 (2011).
- 154 . Synthesis and biological activity of AM-112 and related oxapenem analogues. J. Antibiot. 56(10), 838–847 (2003).
- 155 4-substituted trinems as broad spectrum β-lactamase inhibitors: structure-based design, synthesis, and biological activity. J. Med. Chem. 50(17), 4113–4121 (2007).
- 156 Novel natural product 5,5-trans-lactone inhibitors of human α-thrombin: mechanism of action and structural studies. Biochemistry 37(19), 6645–6657 (1998).
- 157 . Action of strigolactones in plants. Enzymes 35, 57–84 (2014).
- 158 Kinetics of avibactam inhibition against Class A, C, and D β-lactamases. J. Biol. Chem. 288(39), 27960–27971 (2013). • Presents detailed kinetic analyses on avibactam with serine β-lactamases.
- 159 . Inactivation of TEM-1 by avibactam (NXL-104): insights from quantum mechanics/molecular mechanics metadynamics simulations. Biochemistry 53(31), 5174–5185 (2014).
- 160 Mechanistic studies of the inactivation of TEM-1 and P99 by NXL104, a novel non-β-lactam β-lactamase inhibitor. Antimicrob. Agents Chemotherap. 54(12), 5132–5138 (2010).
- 161 Structural insight into potent broad-spectrum inhibition with reversible recyclization mechanism: avibactam in complex with CTX-M-15 and Pseudomonas aeruginosa AmpC β-lactamases. Antimicrob. Agents Chemotherap. 57(6), 2496–2505 (2013).
- 162 Molecular basis of selective inhibition and slow reversibility of avibactam against class D carbapenemases: a structure-guided study of OXA-24 and OXA-48. ACS Chem. Biol. 10(2), 591–600 (2015).
- 163 In vitro activity of AVE1330A, an innovative broad-spectrum non-β-lactam β-lactamase inhibitor. J. Antimicrob. Chemother. 54(2), 410–417 (2004).
- 164 . Investigations on recyclisation and hydrolysis in avibactam mediated serine β-lactamase inhibition. Org. Biomol. Chem. 14(17), 4116–4128 (2016).
- 165 . Structural and biochemical characterization of the interaction between KPC-2 β-lactamase and β-lactamase inhibitor protein. Biochemistry 48(39), 9185–9193 (2009). •• Highlights the interactions between KPC-2 and avibactam.
- 166 Ceftazidime-avibactam: a novel cephalosporin/β-lactamase inhibitor combination. Drugs 73(2), 159–177 (2013).
- 167 Activities of ceftazidime, ceftaroline, and aztreonam alone and combined with avibactam against isogenic Escherichia coli strains expressing selected single β-lactamases. Diagn. Microbiol. Infect. Dis. 82(1), 65–69 (2015).
- 168 . NXL104 combinations versus Enterobacteriaceae with CTX-M extended-spectrum β-lactamases and carbapenemases. J. Antimicrob. Chemother. 62(5), 1053–1056 (2008).
- 169 . Activity of ceftaroline-avibactam tested against Gram-negative organism populations, including strains expressing one or more β-lactamases and methicillin-resistant Staphylococcus aureus carrying various staphylococcal cassette chromosome mec types. Antimicrob. Agents Chemother. 56(9), 4779–4785 (2012).
- 170 . Activity of chequerboard combinations of ceftaroline and NXL104 versus β-lactamase-producing Enterobacteriaceae. J. Antimicrob. Chemother. 65(7), 1428–1432 (2010).
- 171 In vitro activities of ceftazidime-avibactam and aztreonam-avibactam against 372 Gram-negative bacilli collected in 2011 and 2012 from 11 teaching hospitals in China. Antimicrob. Agents Chemother. 58(3), 1774–1778 (2014).
- 172 . Carbapenemases: the versatile β-lactamases. Clin. Microbiol. Rev. 20(3), 440–458, table of contents (2007).
- 173 Discovery of MK-7655, a β-lactamase inhibitor for combination with Primaxin(R). Bioorg. Med. Chem. Lett. 24(3), 780–785 (2014).
- 174 In vitro and in vivo activities of OP0595, a new diazabicyclooctane, against CTX-M-15-positive Escherichia coli and KPC-positive Klebsiella pneumoniae. Antimicrob. Agents Chemother. 60(5), 3001–3006 (2016).
- 175 . Activity of OP0595/β-lactam combinations against Gram-negative bacteria with extended-spectrum, AmpC and carbapenem-hydrolysing β-lactamases. J. Antimicrob. Chemother. 70(11), 3032–3041 (2015).
- 176 . Interactions of OP0595, a novel triple-action diazabicyclooctane, with β-lactams against OP0595-resistant Enterobacteriaceae mutants. Antimicrob. Agents Chemother. 60(1), 554–560 (2015).
- 177 . In vitro activity of MK-7655, a novel β-lactamase inhibitor, in combination with imipenem against carbapenem-resistant Gram-negative bacteria. Antimicrob. Agents Chemother. 56(7), 3753–3757 (2012).
- 178 . Activity of MK-7655 combined with imipenem against Enterobacteriaceae and Pseudomonas aeruginosa. J. Antimicrob. Chemother. 68(10), 2286–2290 (2013).
- 179 . In vitro and in vivo activities of AM-112, a novel oxapenem. Antimicrob. Agents Chemother. 47(5), 1652–1657 (2003).
- 180 Alkylidene oxapenem β-lactamase inhibitors revisited: potent broad spectrum activity but new stability challenges. ACS Med. Chem. Lett. 5(8), 915–920 (2014).
- 181 . Activity of biapenem (RPX2003) combined with the boronate β-lactamase inhibitor RPX7009 against carbapenem-resistant Enterobacteriaceae. J. Antimicrob. Chemother. 68(8), 1825–1831 (2013).


