







Inhaled nitric oxide therapy for extrapulmonary inflammation
Abstract
Inhaled nitric oxide (iNO) has been used extensively to treat pulmonary hypertension primarily in newborns. This therapy is a safe and effective therapy to improve the matching between airway ventilation and blood oxygenation. A key conceptual component of iNO therapy is that effects are limited to the pulmonary compartment thereby avoiding unwanted systemic effects. The mechanism underlying this model is that any NO entering the blood stream is rapidly oxidized to nitrate, a relatively inert anion that is excreted. Mediating this oxidation is oxyhemoglobin that becomes oxidized to methemoglobin, accumulation of which is limited by erythrocyte methemoglobin reductase. In this article, we discuss studies that dismiss the notion that once in the blood stream iNO is inactivated and show that a surprising result of iNO therapy is the formation of stable NO-derived products that circulate and can elicit NO-dependent signaling in extra-pulmonary tissues. This pathway has the potential to open up new applications for iNO for treatment of systemic diseases associated with loss of NO signaling.
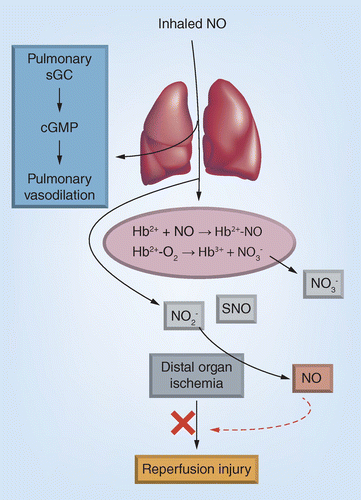
Inhaled nitric oxide (iNO) dilates the pulmonary vasculature by binding to and activating soluble guanylate cyclasae. Any iNO that enters the circulation rapidly reacts with erythrocytic oxyhemoglobin or deoxyhemoglobin forming nitrate and nitrosylhemoglobin, respectively. However, also produced is nitrite and in experimental models, S-nitrosothiols also, but the mechanisms of iNO oxidation to these remains unclear. Nitrite circulates and is reduced back to NO in ischemic tissues by oxygen-sensitive mechanisms, which protects against inflammatory injury. In this paradigm, nitrite-dependent NO formation is targeted to the extrapulmonary tissues under stress.
Inhaled NO therapy for pulmonary hypertension
Nitrogen monoxide (also referred to as nitric oxide [NO]), is a diatomic gas and free radical that has pleiotropic functions in mammals. Loss or dysfunctional NO signaling leads to the development and propagation of various diseases, especially those associated with inflammation. This understanding has led to the testing of NO repletion strategies. Perhaps the most widely used to date are phosphodiesterase-5 (PDE5) inhibitors which inhibit degradation of 3′,5′-cyclic guanosine monophosphate (cGMP) and thereby increase the signaling efficacy for a given amount of NO (Box 1) [1]. Such indirect approaches to modulate NO-signaling cascades are clearly successful. However, NO-formation or delivery strategies are still needed especially in cases where endogenous NO formation is compromised. While many NO-donor or NO-releasing compounds have been synthesized and used widely in the research setting, clinical use is limited by unwanted systemic effects including hypotension [2]. Therefore, delivery of NO in tissue-targeted fashion is key for a successful therapeutic.
With this framework in mind, one of the most successful NO-based therapies, and the only approved therapeutic that employs NO itself (as a gas), has been inhaled nitric oxide (iNO) for the treatment of pulmonary hypertension in the newborn, with applications to adult pulmonary hypertension, lung transplantation and platelet aggregation also reported [3–7]. Abnormal lung or heart development, procedures to correct congenital heart disease and a host of indications associated with prematurity can lead to life-threatening pulmonary hypertension. In the setting of hypertension due to pulmonary vasoconstriction iNO therapy dilates the pulmonary circulation and importantly, does so in areas where the airways are open, thereby effectively improving ventilation–perfusion matching.
NO gas can be toxic however, via secondary generation of reactive nitrogen species including nitrogen dioxide (Box 2) [8], and as mentioned above, any NO-based therapy has to contend with limiting unwanted systemic effects and ensuring that methemoglobinemia does not occur. Effective inhalation protocols that minimize nitrogen dioxide formation (<1 ppm) have been developed, and unless there is a deficiency in methemoglobin reductase, clinically significant methemoglobinemia is not common. With respect to tissue targeting, the concept behind iNO was that inhalation administration would replete NO directly where needed (i.e., the pulmonary circulation) [3]. Systemic toxicity is limited by virtue of the fact that any NO that transits across the pulmonary vascular wall would be immediately and efficiently inactivated by its oxidation to nitrate mediated by erythrocytic hemoglobin (Figure 1). This therapeutic model is supported by excellent clinical efficacy and a track record confirming safety. However, as discussed herein, it is now evident that iNO-dependent modulation of NO signaling is not limited to the pulmonary circulation and that iNO-derived products in the circulation, while not toxic, have the potential to also improve NO bioavailability in systemic tissues.
Inhaled nitric oxide & extrapulmonary effects
The first evidence that iNO could have systemic effects was obtained in experimental models of ischemia-reperfusion injury, in the feline mesenteric circulation [9]. Inhibition of local NO production from nitric oxide synthase or ischemia-reperfusion stimulated an acute inflammatory response indexed by increased binding of leukocytes to the vascular endothelium. Pre-emptive administration to iNO, and specifically before the reperfusion period, resulted in attenuated inflammatory tissue injury. This seminal finding has been reproduced in multiple laboratories and importantly in different experimental models of ischemia-reperfusion injury where decreased NO bioavailability plays a role in the pathology [10–15]. This includes inflammatory injury in the heart, lung and brain with therapeutic efficacy of iNO in preserving cellular function demonstrated. Importantly, at the doses tested, iNO had no effect on systemic hemodynamics and only appears to affect function in tissues where a deficit in endogenous NO signaling is apparent.
Extension of these results into human models confirmed the potential for extrapulmonary effects of iNO. iNO improved local forearm blood flow in healthy humans treated with nitric oxide synthase inhibitors and increased exercise induced blood flow also. No effect of iNO was apparent in the forearm where endogenous NO formation remained intact [16]. Moreover, compared with placebo, pre-emptive iNO administration to liver transplant patients improved the rate of allograft function recovery post-transplantation, consistent with a lesser extent of ischemia-reperfusion injury in patients receiving the iNO [17]. Additional evidence for iNO-mediated protection against inflammation in humans has been reported in the context of tourniquet induced ischemic stress in knee surgery patients [18] and cardiac surgery patients [19]. Collectively these studies have also defined therapeutic parameters when considering iNO as a therapeutic to improve NO signaling in extrapulmonary tissues. Specifically, the effects are only evident at the higher doses of iNO (40–80 ppm) used for pulmonary hypertension treatment. Lower doses of iNO (5–20 ppm) that are efficacious for the latter do not show any effect for systemic responses. Moreover, the efficacy is only observed if iNO is administered prior to the stress that leads to loss of endogenous NO signaling. As described below, these characteristics have been important in elucidating the mechanisms by which iNO affects systemic NO bioavailability.
Mechanisms of iNO systemic effects
How iNO repletes NO at distal sites remains to be determined, but candidate mechanisms have been proposed. NO dissolved in solution in the blood has a half-life of milliseconds due to reaction with hemoglobin (Box 3). This property excludes direct transport of solvated NO from the lungs to the periphery as a viable mechanism. The generally accepted paradigm is that inhalation of NO results in the formation of a relatively more stable intermediate that can circulate, and which can mediate NO signaling. Another required element of this mechanism informed by the therapeutic studies is that this intermediate elicits NO signaling only in areas where endogenous NO signaling is compromised, and does not elicit NO-dependent effects everywhere. Interestingly, tissue hypoxia or ischemic stress is a common feature of models/diseases in which iNO has been shown to improve NO signaling suggesting that lower oxygen tensions is one mechanism by which targeting is achieved.
As noted above, the predominant fate of iNO in the circulation is oxidation to nitrate due to hemoglobin mediated oxidation. For example, in liver transplant patients, iNO (80 ppm) increases plasma nitrate at a rate of approximately 0.01–0.03 uM/min, that continues for the duration of the iNO exposure. However, mammalian systems are not known to express efficient reductases that can reduce nitrate ultimately to NO. We note that nitrate is reduced to nitrite by oral cavity based nitrate reductase expressing bacteria, and perhaps by xanthine oxidoreductase [20], but this occurs relatively slowly (hours) compared with iNO effects, which are seen immediately upon induction of stress.
A more viable candidate is nitrite. iNO exposure increases circulating nitrite levels which reach a steady state; in liver transplant patients steady state nitrite concentrations are approximately 1–2 µM for iNO at 80 ppm. This concentration is an order of magnitude lower than nitrate and the steady state of nitrite reflects a dynamic balance between formation and loss. The latter is likely ferrous hemoglobin mediated oxidation to nitrate. How nitrite is formed from iNO however is less understood. It has been demonstrated that ceruloplasmin, a copper containing enzyme, can compete with erythrocytic hemoglobin to oxidize NO to nitrite in the blood [21]. It is likely that ceruloplasmin or another similar metalloprotein either in the circulation or in the lung mediate iNO oxidation to nitrite. Contrary to nitrate, nitrite reduction to NO by one-electron is a facile process in mammals and can be mediated by several metallo/- heme proteins [20]. Also, nitrite reduction to NO is faster in hypoxic tissues and nitrite therapy prevents ischemia-reperfusion injury in multiple organs [22]. The parallels between systemic effects of iNO and nitrite underlie the working model that the latter is formed by iNO, circulates and replenishes NO signaling in extrapulmonary tissues (Figure 1).
An alternative and complementary model invokes a role for S-nitrosothiols as the products formed from iNO which then mediate systemic effects. S-nitrosothiols can stimulate various NO-dependent processes, although the hypoxia sensitivity is less evident compared with nitrite. Evidence for this mechanism is provided by measured increases in plasma S-nitrosothiols (of albumin) in cats exposed to iNO [23]. However, similar increases are not observed in humans exposed to iNO [16,17].
Conclusion
Inhaled nitric oxide is a well established therapeutic for pulmonary hypertension in the setting of vasoconstriction. Observations of the benefit of iNO in animal and human models of ischemia-reperfusion injury led to the appreciation of the therapeutic potential of iNO to replenish NO signaling in extrapulmonary tissues. Mechanistic insights have focused on soluble, circulating species formed during iNO administration such as nitrite, nitrate and S-nitrosothiols with a role for the first candidate supported by similar therapeutic profiles between and iNO and exogenous administration of nitrite.
Nitric oxide–cGMP signaling cascade.
• The classic NO-signaling cascade involves formation from one of three nitric oxide synthase isoforms (NOS 1, NOS 2, NOS 3) and subsequent activation of the enzyme soluble guanylate cyclase, which in turn catalyzes the formation of 3′,5′-cyclic guanosine monophosphate (cGMP) from GTP. cGMP controls multiple cellular functions, in part by activating PKG. The levels of cGMP are controlled by its formation and degradation, the latter by phosphodiesterase-5 mediated hydrolysis. Inhibition of phosphodiesterase-5 (by sildenafil for example) is a widely used therapeutic approach to improve NO-dependent signaling
Nitric oxide oxidation to nitrogen dioxide.
• In the gas phase, nitric oxide reacts with oxygen to form nitrogen dioxide according to the following equation:

• NO2 is a brown toxic gas able to oxidize and nitrate biomolecules leading to cell damage and death
Nitric oxide metabolism by red blood cells.
• NO reacts with the ferrous heme of both oxyhemoglobin (Fe2+-O2) or deoxyhemoglobin (Fe2+) forming methemoglobin (Fe3+) or nitrosylhemoglobin (Fe2+-NO) respectively. Both these reactions are rapid (rate constants being 2–4 × 107M-1s-1), which coupled with the high heme concentrations in blood (˜10 mM) in blood, resulting in the short half-life for NO in the intravascular compartment

Executive summary
Loss or dysfunctional NO signaling leads to the development and propagation of various diseases, especially those associated with inflammation.
With this framework in mind, one of the most successful NO-based therapies, and the only approved therapeutic that employs NO itself (as a gas), has been inhaled nitric oxide (iNO) for the treatment of pulmonary hypertension in the newborn.
The first evidence that iNO could have systemic effects were obtained in experimental models of ischemia-reperfusion injury, in the feline mesenteric circulation.
Pre-emptive administration to iNO, and specifically before the reperfusion period, resulted in attenuated inflammatory tissue injury.
Importantly, at the doses tested, iNO had no effect on systemic hemodynamics and only appears to affect function in tissues where a deficit in endogenous NO signaling is apparent.
The generally accepted paradigm is that inhalation of NO results in the formation of a relatively more stable intermediate that can circulate, and which can mediate NO signaling.
Financial & competing interests disclosure
RP Patel is a coinventor on a patent for use of nitrite salts for the treatment of cardiovascular conditions. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
Open access
This work is licensed under a Creative Commons Attribution 4.0 License. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
References
- 1 . Effectiveness and safety of phosphodiesterase 5 inhibitors in patients with cardiovascular disease and hypertension. Curr. Hypertens. Rep. 15(5), 475–483 (2013).
- 2 . S-Nitrosothiol biology and therapeutic potential in metabolic disease. Curr. Opin. Investig. Drugs 11(10), 1127–1134 (2010).
- 3 . Inhaled NO as a therapeutic agent. Cardiovasc. Res. 75(2), 339–348 (2007).
- 4 . Bench-to-bedside review: inhaled nitric oxide therapy in adults. Crit. Care 13(3), 221 (2009).
- 5 . Inhaled nitric oxide: clinical applications, indications, and toxicology. Can. J. Anaesth. 44(9), 973–988 (1997).
- 6 Inhaled nitric oxide attenuates reperfusion injury in non-heartbeating-donor lung transplantation. Paris-Sud University Lung Transplantation Group. Transplantation 63(10), 1380–1386 (1997).
- 7 Randomized, placebo-controlled, blinded and cross-matched study on the antiplatelet effect of inhaled nitric oxide in healthy volunteers. Thromb. Haemost. 83(2), 309–315 (2000).
- 8 . Delivery and monitoring of inhaled nitric oxide in patients with pulmonary hypertension. Crit. Care Med. 22(6), 930–938 (1994).
- 9 Inhaled NO as a viable antiadhesive therapy for ischemia/reperfusion injury of distal microvascular beds. J. Clin. Invest. 101(11), 2497–2505 (1998).
- 10 . Inhaled NO impacts vascular but not extravascular compartments in postischemic peripheral organs. Am. J. Physiol. 277(2 Pt 2), H676–H682 (1999).
- 11 Inhaled nitric oxide attenuates apoptosis in ischemia-reperfusion injury of the rabbit lung. Ann. Thorac. Surg. 78(1), 292–297 (2004).
- 12 . Effects of inhaled nitric oxide on lung injury after intestinal ischemia-reperfusion in rats. Shock 23(2), 150–155 (2005).
- 13 Inhaled nitric oxide decreases infarction size and improves left ventricular function in a murine model of myocardial ischemia-reperfusion injury. Am. J. Physiol. Heart Circ. Physiol. 291(1), H379–H384 (2006).
- 14 Inhalation of nitric oxide prevents ischemic brain damage in experimental stroke by selective dilatation of collateral arterioles. Circ. Res. 110(5), 727–738 (2012).
- 15 Adverse effects of hemorrhagic shock resuscitation with stored blood are ameliorated by inhaled nitric oxide in lambs*. Crit. Care Med. 41(11), 2492–2501 (2013).
- 16 Effects of inhaled nitric oxide on regional blood flow are consistent with intravascular nitric oxide delivery. J. Clin. Invest. 108(2), 279–287 (2001).
- 17 Inhaled NO accelerates restoration of liver function in adults following orthotopic liver transplantation. J. Clin. Invest. 117(9), 2583–2591 (2007).
- 18 . Inhaled nitric oxide attenuates reperfusion inflammatory responses in humans. Anesthesiology 106(2), 275–282 (2007).
- 19 Supplemental nitric oxide and its effect on myocardial injury and function in patients undergoing cardiac surgery with extracorporeal circulation. J. Thorac. Cardiovasc. Surg. 127(1), 44–50 (2004).
- 20 Nitrate and nitrite in biology, nutrition and therapeutics. Nat. Chem. Biol. 5(12), 865–869 (2009).
- 21 Ceruloplasmin is a NO oxidase and nitrite synthase that determines endocrine NO homeostasis. Nat. Chem. Biol. 2(9), 486–493 (2006).
- 22 Cytoprotective effects of nitrite during in vivo ischemia-reperfusion of the heart and liver. J. Clin. Invest. 115(5), 1232–1240 (2005).
- 23 Enhanced S-nitroso-albumin formation from inhaled NO during ischemia/reperfusion. Circ. Res. 94(4), 559–565 (2004).


